 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1spg: CARBONMONOXY HEMOGLOBIN FROM THE TELEOST FISH LEIOSTOMUS XANTHURUS -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1spg | ||||||
|---|---|---|---|---|---|---|---|
| Title | CARBONMONOXY HEMOGLOBIN FROM THE TELEOST FISH LEIOSTOMUS XANTHURUS | ||||||
 Components Components | (HEMOGLOBIN) x 2 | ||||||
 Keywords Keywords | OXYGEN TRANSPORT / CARBON MONOXIDE / R-STATE / LEIOSTOMUS XANTHURUS / TELEOST FISH / ROOT EFFECT / GLOBIN | ||||||
| Function / homology |  Function and homology information Function and homology informationhaptoglobin binding / organic acid binding / haptoglobin-hemoglobin complex / hemoglobin complex / hydrogen peroxide catabolic process / oxygen carrier activity / peroxidase activity / oxygen binding / blood microparticle / heme binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Leiostomus xanthurus (spot croaker) Leiostomus xanthurus (spot croaker) | ||||||
| Method |  X-RAY DIFFRACTION / INITIAL PHASES FROM THE ISOMORPHOUS PHOSPHATE BOUND SPOT R-STATE HEMOGLOBIN (S. MYLVAGANAM, E. GETZOFF, UNPUBLISHED) / Resolution: 1.95 Å X-RAY DIFFRACTION / INITIAL PHASES FROM THE ISOMORPHOUS PHOSPHATE BOUND SPOT R-STATE HEMOGLOBIN (S. MYLVAGANAM, E. GETZOFF, UNPUBLISHED) / Resolution: 1.95 Å | ||||||
 Authors Authors | Mylvaganam, S.E. / Getzoff, E.D. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1996 Title: Structural basis for the root effect in haemoglobin. Authors: Mylvaganam, S.E. / Bonaventura, C. / Bonaventura, J. / Getzoff, E.D. #1: Journal: J.Biol.Chem. / Year: 1976Title: Spot Hemoglobin. Studies on the Root Effect Hemoglobin of a Marine Teleost Authors: Bonaventura, C. / Sullivan, B. / Bonaventura, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1spg.cif.gz | 76.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1spg.ent.gz | 55.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1spg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sp/1spgftp://data.pdbj.org/pub/pdb/validation_reports/sp/1spg | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|















-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THERE IS AN ALPHA-BETA DIMER IN THE ASYMMETRIC UNIT. THE ALPHA-BETA DIMERS ARE RELATED BY A CRYSTALLOGRAPHIC TWO-FOLD THAT IS COINCIDENT WITH THE MOLECULAR TWO-FOLD OF THE ALPHA2BETA2 TETRAMER. |
-Components
| #1: Protein | Mass: 15684.213 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: R-STATE, CARBONMONOXY BOUND ROOT EFFECT HEMOGLOBIN / Source: (natural) Leiostomus xanthurus (spot croaker) / Tissue: BLOOD / References: UniProt: P56250 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 16316.785 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: R-STATE, CARBONMONOXY BOUND ROOT EFFECT HEMOGLOBIN / Source: (natural) Leiostomus xanthurus (spot croaker) / Tissue: BLOOD / References: UniProt: P56251 | ||||||
| #3: Chemical |   Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4#4: Chemical | 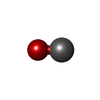  Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 175 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 175 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 40 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 | ||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293.15 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ELLIOTT GX-21 / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Nov 29, 1993 |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 1.95 Å / Num. obs: 16125 / % possible obs: 76 % / Observed criterion σ(I): 0 / Redundancy: 2 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 26.6 |
| Reflection shell | Resolution: 1.95→2.07 Å / % possible all: 38 |
| Reflection | *PLUS Num. measured all: 27397 |
| Reflection shell | *PLUS % possible obs: 38 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: INITIAL PHASES FROM THE ISOMORPHOUS PHOSPHATE BOUND SPOT R-STATE HEMOGLOBIN (S. MYLVAGANAM, E. GETZOFF, UNPUBLISHED) Resolution: 1.95→10 Å / σ(F): 4
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
