 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1shv | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF SHV-1 BETA-LACTAMASE | ||||||
 Components Components | PROTEIN (BETA-LACTAMASE SHV-1) | ||||||
 Keywords Keywords | HYDROLASE / BETA-LACTAM HYDROLASE / PENICILLINASE / DETERGENT BINDING / DRUG DESIGN | ||||||
| Function / homology |  Function and homology information Function and homology informationbeta-lactam antibiotic catabolic process / beta-lactamase / beta-lactamase activity / response to antibiotic Similarity search - Function | ||||||
| Biological species |  Klebsiella pneumoniae (bacteria) Klebsiella pneumoniae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.98 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.98 Å | ||||||
 Authors Authors | Kuzin, A.P. / Nukaga, M. / Nukaga, Y. / Hujer, A. / Bonomo, R.A. / Knox, J.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Structure of the SHV-1 beta-lactamase. Authors: Kuzin, A.P. / Nukaga, M. / Nukaga, Y. / Hujer, A.M. / Bonomo, R.A. / Knox, J.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1shv.cif.gz | 65.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1shv.ent.gz | 47.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1shv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sh/1shvftp://data.pdbj.org/pub/pdb/validation_reports/sh/1shv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1xpbS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28907.018 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: 1 DETERGENT MOLECULE (CYMAL-6) BOUND NON-COVALENTLY Source: (gene. exp.) Klebsiella pneumoniae (bacteria) / Strain: 15571 / Gene: BLA / Plasmid: PBCSK / Species (production host): Escherichia coli / Gene (production host): BLAProduction host: Strain (production host): DH10B References: UniProt: P14557, UniProt: P0AD64*PLUS, beta-lactamase | ||
|---|---|---|---|
| #2: Chemical | ChemComp-MA4 / 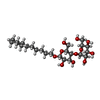  Mass: 508.600 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H44O11 Mass: 508.600 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H44O11 | ||
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 81 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 81 / Source method: isolated from a natural source / Formula: H2O | ||
| Has protein modification | Y | ||
| Nonpolymer details | DETERGENT USED IN CRYSTALLIZ| Sequence details | WE USE A CONSENSUS NUMBERING IN WHICH 239 AND 253 ARE NOT USED FOR THIS ENZYME. HERE, 238 AND 240 ...WE USE A CONSENSUS NUMBERING IN WHICH 239 AND 253 ARE NOT USED FOR THIS ENZYME. HERE, 238 AND 240 ARE LINKED, AS ARE 252 AND 254. TER ARG: NUMBERING FOR THE MATURE PROTEIN STARTS AT 26 (USING A CONSENSUS CONVENTION | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.08 Å3/Da / Density % sol: 41 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 Details: VAPOR DIFFUSION, WITH 2MG/ML PROTEIN IN 15% PEG6000, 50MM HEPES PH7.0 0.65MM CYMAL-6, OVER 30% PEG RESERVOIR AT ROOM TEMPERATURE | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Oct 1, 1998 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.96→43 Å / Num. obs: 49725 / % possible obs: 0.85 % / Redundancy: 3.3 % / Rsym value: 0.065 / Net I/σ(I): 15.5 |
| Reflection shell | Resolution: 1.96→2.09 Å / Redundancy: 1.95 % / Mean I/σ(I) obs: 3.2 / Rsym value: 0.194 / % possible all: 0.4 |
| Reflection | *PLUS Num. obs: 15082 / % possible obs: 85 % / Num. measured all: 49725 / Rmerge(I) obs: 0.065 |
| Reflection shell | *PLUS % possible obs: 40 % / Num. unique obs: 1156 / Num. measured obs: 2259 / Rmerge(I) obs: 0.194 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1XPB Resolution: 1.98→8 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0 / Details: NO IDEAL GEOMETRY FOR DETERGENT MOLECULE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.205 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.98→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.98→2.06 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.182 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
