 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1qqs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | NEUTROPHIL GELATINASE ASSOCIATED LIPOCALIN HOMODIMER | |||||||||
 Components Components | NEUTROPHIL GELATINASE | |||||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / NEUTROPHIL LIPOCALIN / SIGNAL PROTEIN / GLYCOPROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of iron ion import across plasma membrane / positive regulation of hippocampal neuron apoptotic process / positive regulation of endothelial tube morphogenesis / negative regulation of hippocampal neuron apoptotic process / positive regulation of cell projection organization / Metal sequestration by antimicrobial proteins / response to mycotoxin / response to kainic acid / siderophore transport / cellular response to increased oxygen levels ...positive regulation of iron ion import across plasma membrane / positive regulation of hippocampal neuron apoptotic process / positive regulation of endothelial tube morphogenesis / negative regulation of hippocampal neuron apoptotic process / positive regulation of cell projection organization / Metal sequestration by antimicrobial proteins / response to mycotoxin / response to kainic acid / siderophore transport / cellular response to increased oxygen levels / response to blue light / cellular response to X-ray / response to fructose / short-term memory / cellular response to interleukin-6 / enterobactin binding / iron ion sequestering activity / response to herbicide / response to iron(II) ion / positive regulation of reactive oxygen species biosynthetic process / cellular response to interleukin-1 / long-term memory / extrinsic apoptotic signaling pathway in absence of ligand / positive regulation of endothelial cell migration / cellular response to nutrient levels / acute-phase response / cellular response to nerve growth factor stimulus / Iron uptake and transport / specific granule lumen / cellular response to tumor necrosis factor / cellular response to amyloid-beta / response to virus / cellular response to hydrogen peroxide / positive regulation of cold-induced thermogenesis / cellular response to lipopolysaccharide / protease binding / Interleukin-4 and Interleukin-13 signaling / cellular response to hypoxia / defense response to bacterium / iron ion binding / response to xenobiotic stimulus / innate immune response / Neutrophil degranulation / positive regulation of gene expression / : / extracellular exosome / extracellular region / identical protein binding Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.4 Å X-RAY DIFFRACTION / Resolution: 2.4 Å | |||||||||
 Authors Authors | Goetz, D.H. / Willie, S.T. / Armen, R. / Bratt, T. / Borregaard, N. / Strong, R.K. | |||||||||
 Citation Citation | Journal: Biochemistry / Year: 2000 Title: Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin Authors: Goetz, D.H. / Willie, S.T. / Armen, R.S. / Bratt, T. / Borregaard, N. / Strong, R.K. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1qqs.cif.gz | 52.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1qqs.ent.gz | 36.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1qqs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qq/1qqsftp://data.pdbj.org/pub/pdb/validation_reports/qq/1qqs | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 20012.865 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  unidentified baculovirus / References: UniProt: P80188 unidentified baculovirus / References: UniProt: P80188 |
|---|---|
| #2: Polysaccharide | alpha-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta- ...alpha-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source |
| #3: Chemical | ChemComp-DKA / 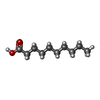  Mass: 172.265 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H20O2 Mass: 172.265 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H20O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.2 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 4.5 Details: 14 % W/W PEG 8K 15% V/V GLYCEROL 50MM ACETATE, pH 4.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 200 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Jun 25, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. all: 7633 / Num. obs: 6110 / % possible obs: 80 % / Observed criterion σ(I): 0 / Redundancy: 6.48 % / Biso Wilson estimate: 18.5 Å2 / Rmerge(I) obs: 0.052 / Net I/σ(I): 10.8 |
| Reflection shell | Resolution: 2.4→2.44 Å / Redundancy: 1.04 % / Rmerge(I) obs: 0.019 / % possible all: 38.7 |
| Reflection | *PLUS Num. obs: 6872 / % possible obs: 88.1 % |
| Reflection shell | *PLUS % possible obs: 38.7 % / Rmerge(I) obs: 0.109 / Mean I/σ(I) obs: 5.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.4→20 Å / Rfactor Rfree error: 0.011 / Data cutoff high absF: 290473.57 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH AND HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 37.25 Å2 / ksol: 0.3404 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.052 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 10.9 % / Rfactor obs: 0.216 / Rfactor Rfree: 0.285 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 24.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.424 / % reflection Rfree: 13.5 % / Rfactor Rwork: 0.298 |
