 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1q84: Crystal structure of the mouse acetylcholinesterase-TZ2PA6 anti c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1q84 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the mouse acetylcholinesterase-TZ2PA6 anti complex | ||||||
 Components Components | Acetylcholinesterase | ||||||
 Keywords Keywords | HYDROLASE / SERINE ESTERASE / ACETYLCHOLINESTERASE / bifunctional inhibitor | ||||||
| Function / homology |  Function and homology information Function and homology informationacetylcholine metabolic process / serine hydrolase activity / acetylcholine catabolic process / acetylcholinesterase / cholinesterase activity / positive regulation of dendrite morphogenesis / acetylcholine receptor signaling pathway / osteoblast development / choline metabolic process / acetylcholine binding ...acetylcholine metabolic process / serine hydrolase activity / acetylcholine catabolic process / acetylcholinesterase / cholinesterase activity / positive regulation of dendrite morphogenesis / acetylcholine receptor signaling pathway / osteoblast development / choline metabolic process / acetylcholine binding / positive regulation of axonogenesis / acetylcholinesterase activity / retina development in camera-type eye / regulation of receptor recycling / basement membrane / side of membrane / synaptic cleft / collagen binding / laminin binding / synapse assembly / neuromuscular junction / receptor internalization / response to insulin / nuclear envelope / positive regulation of cold-induced thermogenesis / presynaptic membrane / postsynaptic membrane / cell adhesion / membrane raft / endoplasmic reticulum lumen / axon / hydrolase activity / neuronal cell body / synapse / dendrite / perinuclear region of cytoplasm / Golgi apparatus / cell surface / protein homodimerization activity / : / extracellular region / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.45 Å | ||||||
 Authors Authors | Bourne, Y. / Kolb, H.C. / Radic, Z. / Sharpless, K.B. / Taylor, P. / Marchot, P. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.Usa / Year: 2004 Title: Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Authors: Bourne, Y. / Kolb, H.C. / Radic, Z. / Sharpless, K.B. / Taylor, P. / Marchot, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1q84.cif.gz | 229.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1q84.ent.gz | 181.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1q84.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q8/1q84ftp://data.pdbj.org/pub/pdb/validation_reports/q8/1q84 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1q83C  1j06S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |























-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / Refine code: 3
|
-Components
| #1: Protein | Mass: 63800.230 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  Homo sapiens (human) / References: UniProt: P21836, acetylcholinesterase Homo sapiens (human) / References: UniProt: P21836, acetylcholinesterase#2: Sugar |   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Chemical | ChemComp-P6G / | 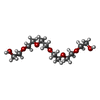  Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM Mass: 282.331 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H26O7 / Comment: precipitant*YM#4: Chemical |   Mass: 661.860 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C42H45N8 Mass: 661.860 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C42H45N8#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 338 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 338 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.94 Å3/Da / Density % sol: 68.78 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6.75 Details: 25-32% PEG 600, 20-100 mM Hepes, pH 6.75, VAPOR DIFFUSION, HANGING DROP, temperature 277K | ||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop / PH range low: 7.5 / PH range high: 6 | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 Å / Beamline: ID14-2 / Wavelength: 0.933 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Dec 5, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.45→20 Å / Num. obs: 74834 / % possible obs: 99.8 % / Redundancy: 3.9 % / Biso Wilson estimate: 57 Å2 / Rsym value: 0.057 / Net I/σ(I): 9.3 |
| Reflection | *PLUS Lowest resolution: 25 Å / Num. measured all: 549476 / Rmerge(I) obs: 0.057 |
| Reflection shell | *PLUS % possible obs: 99.7 % / Rmerge(I) obs: 0.435 / Mean I/σ(I) obs: 1.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 1J06 Resolution: 2.45→20 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.94 / SU B: 6.013 / SU ML: 0.134 / Cross valid method: THROUGHOUT / ESU R: 0.219 / ESU R Free: 0.183 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.036 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.45→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.45→2.513 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 25 Å / % reflection Rfree: 2 % / Rfactor Rfree: 0.214 / Rfactor Rwork: 0.184 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
