 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nz0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | RNASE P PROTEIN FROM THERMOTOGA MARITIMA | ||||||
 Components Components | Ribonuclease P protein component | ||||||
 Keywords Keywords | HYDROLASE / ENDONUCLEASE / RNASE / ALFA-BETA SANDWICH / DIMER / Structural Genomics / BSGC structure funded by NIH / Protein Structure Initiative / PSI / Berkeley Structural Genomics Center | ||||||
| Function / homology |  Function and homology information Function and homology informationribonuclease P complex / 3'-tRNA processing endoribonuclease activity / tRNA 3'-end processing / ribonuclease P / ribonuclease P activity / tRNA 5'-leader removal / tRNA binding Similarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 1.2 Å | ||||||
 Authors Authors | Kazantsev, A.V. / Krivenko, A.A. / Harrington, D.J. / Carter, R.J. / Holbrook, S.R. / Adams, P.D. / Pace, N.R. / Berkeley Structural Genomics Center (BSGC) | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: High-resolution structure of RNase P protein from Thermotoga maritima. Authors: Kazantsev, A.V. / Krivenko, A.A. / Harrington, D.J. / Carter, R.J. / Holbrook, S.R. / Adams, P.D. / Pace, N.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nz0.cif.gz | 235.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nz0.ent.gz | 191.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1nz0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1nz0_validation.pdf.gz | 454.9 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1nz0_full_validation.pdf.gz | 470.1 KB | Display | |
| Data in XML | 1nz0_validation.xml.gz | 26.7 KB | Display | |
| Data in CIF | 1nz0_validation.cif.gz | 39.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nz/1nz0ftp://data.pdbj.org/pub/pdb/validation_reports/nz/1nz0 | HTTPS FTP |
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |









-Links
 PDBj
PDBj
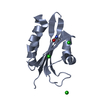
- Assembly
Assembly

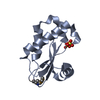

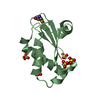










-Components
| #1: Protein | Mass: 14427.935 Da / Num. of mol.: 4 / Mutation: M1S, L41M Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (bacteria) / Gene: RNPA OR TM1463 / Plasmid: PGEX4TA / Production host: #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: SO4#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 533 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 533 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 41.1 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.2 Details: PEG 1500, potassium sulfate, sodium acetate, pH 5.2, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 23 ℃ / Method: vapor diffusion / PH range low: 5.2 / PH range high: 4.8 | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 0.9792 / Wavelength: 0.9792 Å / Beamline: 5.0.2 / Wavelength: 0.9792 / Wavelength: 0.9792 Å |
| Detector | Type: ADSC / Detector: CCD / Date: May 11, 2001 / Details: W16 WIGGLER |
| Radiation | Monochromator: DOUBLE-CRYSTAL SI(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→30 Å / Num. all: 118678 / Num. obs: 118678 / % possible obs: 80 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 5 / Redundancy: 6.53 % / Biso Wilson estimate: 13.688 Å2 / Rmerge(I) obs: 0.059 / Rsym value: 0.059 / Net I/σ(I): 5.3 |
| Reflection shell | Resolution: 1.2→1.35 Å / Redundancy: 4.01 % / Rmerge(I) obs: 0.565 / Mean I/σ(I) obs: 1.1 / Num. unique all: 16982 / Rsym value: 0.565 / % possible all: 38.7 |
| Reflection | *PLUS Num. measured all: 774818 |
| Reflection shell | *PLUS % possible obs: 38.7 % / Num. unique obs: 16982 / Num. measured obs: 67203 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 1.2→30 Å / Num. parameters: 41196 / Num. restraintsaints: 57603 / Isotropic thermal model: anisotropic / Cross valid method: FREE R / Stereochemistry target values: Engh & Huber Details: ANISOTROPIC SCALING APPLIED BY THE METHOD OF PARKIN, MOEZZI & HOPE, J.APPL.CRYST.28(1995)53-56 ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 2.14%
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 148 / Occupancy sum hydrogen: 3913 / Occupancy sum non hydrogen: 4230.3 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.2→30 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL / Version: 97 / Classification: refinement | |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 50 Å / Rfactor Rwork: 0.1634 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
