 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1mrk: STUDIES ON CRYSTAL STRUCTURES ACTIVE CENTER GEOMETRY AND DEPURINE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1mrk | ||||||
|---|---|---|---|---|---|---|---|
| Title | STUDIES ON CRYSTAL STRUCTURES ACTIVE CENTER GEOMETRY AND DEPURINE MECHANISM OF TWO RIBOSOME-INACTIVATING PROTEINS | ||||||
 Components Components | ALPHA-TRICHOSANTHIN | ||||||
 Keywords Keywords | RIBOSOME-INACTIVATING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of defense response to virus / rRNA N-glycosylase / rRNA N-glycosylase activity / defense response / toxin activity / negative regulation of translation Similarity search - Function | ||||||
| Biological species |  Trichosanthes kirilowii (Chinese cucumber) Trichosanthes kirilowii (Chinese cucumber) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.6 Å X-RAY DIFFRACTION / Resolution: 1.6 Å | ||||||
 Authors Authors | Huang, Q. / Liu, S. / Tang, Y. / Jin, S. / Wang, Y. | ||||||
 Citation Citation | Journal: Biochem.J. / Year: 1995 Title: Studies on crystal structures, active-centre geometry and depurinating mechanism of two ribosome-inactivating proteins. Authors: Huang, Q. / Liu, S. / Tang, Y. / Jin, S. / Wang, Y. #1: Journal: Prog.Nat.Sci. / Year: 1994Title: Active Center Geometry and Depurine Mechanism of Two Ribosome-Inactivating Proteins Authors: Tang, Y. / Huang, Q. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1mrk.cif.gz | 76.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1mrk.ent.gz | 58.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1mrk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mr/1mrkftp://data.pdbj.org/pub/pdb/validation_reports/mr/1mrk | HTTPS FTP |
|---|
-Related structure data














-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27166.770 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Trichosanthes kirilowii (Chinese cucumber)References: UniProt: P09989 |
|---|---|
| #2: Chemical | ChemComp-FMC / (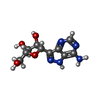  Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 134 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 134 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.51 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 6.1 / Method: vapor diffusion, hanging drop | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Num. obs: 22182 / % possible obs: 70.3 % / Rmerge(I) obs: 0.056 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.6→7 Å / σ(F): 1 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.176 / Rfactor Rwork: 0.176 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_d / Dev ideal: 1.34 |
