 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1k0g | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | THE CRYSTAL STRUCTURE OF AMINODEOXYCHORISMATE SYNTHASE FROM PHOSPHATE GROWN CRYSTALS | ||||||
 要素 要素 | p-aminobenzoate synthase component I | ||||||
 キーワード キーワード | LYASE / AMINODEOXYCHORISMATE SYNTHASE / CHORISMATE / GLUTAMINE / TRYPTOPHAN / PABA SYNTHASE / P-AMINOBENZOATE SYNTHASE | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報aminodeoxychorismate synthase complex / aminodeoxychorismate synthase / tryptophan binding / 4-amino-4-deoxychorismate synthase activity / 4-aminobenzoate biosynthetic process / L-tryptophan biosynthetic process / folic acid biosynthetic process / tetrahydrofolate biosynthetic process / protein heterodimerization activity / magnesium ion binding 類似検索 - 分子機能 | ||||||
| 生物種 |  | ||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 2.05 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 2.05 Å | ||||||
 データ登録者 データ登録者 | Parsons, J.F. / Jensen, P.Y. / Pachikara, A.S. / Howard, A.J. / Eisenstein, E. / Ladner, J.E. | ||||||
 引用 引用 | ジャーナル: Biochemistry / 年: 2002 タイトル: Structure of Escherichia coli aminodeoxychorismate synthase: architectural conservation and diversity in chorismate-utilizing enzymes. 著者: Parsons, J.F. / Jensen, P.Y. / Pachikara, A.S. / Howard, A.J. / Eisenstein, E. / Ladner, J.E. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1k0g.cif.gz | 194.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1k0g.ent.gz | 149.7 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1k0g.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1k0g_validation.pdf.gz | 478.9 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1k0g_full_validation.pdf.gz | 547.9 KB | 表示 | |
| XML形式データ | 1k0g_validation.xml.gz | 45.5 KB | 表示 | |
| CIF形式データ | 1k0g_validation.cif.gz | 64 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/k0/1k0gftp://data.pdbj.org/pub/pdb/validation_reports/k0/1k0g | HTTPS FTP |
-関連構造データ











-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 51022.316 Da / 分子数: 2 / 由来タイプ: 組換発現 / 由来: (組換発現) 参照: UniProt: P05041, 付加脱離酵素(リアーゼ); C-Cリアーゼ; オキソ酸の付加脱離 #2: 化合物 |   分子量: 94.971 Da / 分子数: 2 / 由来タイプ: 合成 / 式: PO4 分子量: 94.971 Da / 分子数: 2 / 由来タイプ: 合成 / 式: PO4#3: 化合物 | 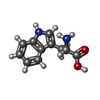  タイプ: L-peptide linking / 分子量: 204.225 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C11H12N2O2 タイプ: L-peptide linking / 分子量: 204.225 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C11H12N2O2#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 560 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 560 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 3.43 Å3/Da / 溶媒含有率: 64.2 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 298 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7.5 詳細: 0.1M HEPES pH7.5, 1.6M Na/K Phosphate, VAPOR DIFFUSION, HANGING DROP, temperature 298K (PROTEIN SOLUTION: 50mM MOPS pH 7 50mM KCL, 5mM MG CL2, 2 mM DTT, 40.2 MG/ML PROTEIN. WELL SOL 0.1 M NA ...詳細: 0.1M HEPES pH7.5, 1.6M Na/K Phosphate, VAPOR DIFFUSION, HANGING DROP, temperature 298K (PROTEIN SOLUTION: 50mM MOPS pH 7 50mM KCL, 5mM MG CL2, 2 mM DTT, 40.2 MG/ML PROTEIN. WELL SOL 0.1 M NA HEPES pH 7.5, 0.8 M NA PHOSPHATE, 0.8 M K PHOSPHATE) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 結晶化 | *PLUS pH: 7.4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 115 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: APS  / ビームライン: 17-ID / 波長: 1.0597 / 波長: 1.06 Å / ビームライン: 17-ID / 波長: 1.0597 / 波長: 1.06 Å | |||||||||
| 検出器 | タイプ: MARRESEARCH / 検出器: CCD / 日付: 2000年9月17日 | |||||||||
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray | |||||||||
| 放射波長 |
| |||||||||
| 反射 | 解像度: 2.05→20 Å / Num. all: 348268 / Num. obs: 88221 / % possible obs: 99.6 % / 冗長度: 4 % / Rmerge(I) obs: 0.063 / Net I/σ(I): 28.1 | |||||||||
| 反射 シェル | 解像度: 2.05→2.16 Å / 冗長度: 4 % / Rmerge(I) obs: 0.327 / Mean I/σ(I) obs: 2.7 / % possible all: 98.6 | |||||||||
| 反射 | *PLUS Num. measured all: 348268 | |||||||||
| 反射 シェル | *PLUS % possible obs: 98.6 % |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: PABB GROWN IN FORMATE, PDB ENTRY 1K0E 解像度: 2.05→10 Å / Num. parameters: 28923 / Num. restraintsaints: 27487 / 交差検証法: FREE R / σ(F): 0 / 立体化学のターゲット値: ENGH AND HUBER 詳細: ANISOTROPIC SCALING APPLIED BY THE METHOD OF PARKIN, MOEZZI & HOPE, J.APPL.CRYST.28(1995)53-56
| |||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: METHOD USED : MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-22 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 7227 | |||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.05→10 Å
| |||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: SHELXL-97 / 分類: refinement | |||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最低解像度: 10 Å / σ(F): 0 / % reflection Rfree: 5.3 % / Rfactor Rwork: 0.173 | |||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | |||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | |||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS タイプ: s_plane_restr / Dev ideal: 0.023 |
