 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ifp: INOVIRUS (FILAMENTOUS BACTERIOPHAGE) STRAIN PF3 MAJOR COAT PROTEI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ifp | ||||||
|---|---|---|---|---|---|---|---|
| Title | INOVIRUS (FILAMENTOUS BACTERIOPHAGE) STRAIN PF3 MAJOR COAT PROTEIN ASSEMBLY | ||||||
 Components Components | MAJOR COAT PROTEIN ASSEMBLY | ||||||
 Keywords Keywords | VIRUS / VIRUS COAT PROTEIN / Helical virus | ||||||
| Function / homology | Inovirus Coat protein B / ATP synthase delta/epsilon subunit, C-terminal domain / Capsid protein G8P / helical viral capsid / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / host cell plasma membrane / Up-down Bundle / Mainly Alpha / Capsid protein G8P Function and homology information Function and homology information | ||||||
| Biological species |  Pseudomonas phage Pf3 (virus) Pseudomonas phage Pf3 (virus) | ||||||
| Method |  FIBER DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å FIBER DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.1 Å | ||||||
 Authors Authors | Welsh, L.C. / Symmons, M.F. / Perham, R.N. / Marvin, D.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1998 Title: Structure of the capsid of Pf3 filamentous phage determined from X-ray fibre diffraction data at 3.1 A resolution. Authors: Welsh, L.C. / Symmons, M.F. / Sturtevant, J.M. / Marvin, D.A. / Perham, R.N. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1995Title: Pf1 Filamentous Bacteriophage: Refinement of a Molecular Model by Simulated Annealing Using 3.3 A Resolution X-Ray Fibre Diffraction Data Authors: Gonzalez, A. / Nave, C. / Marvin, D.A. #2: Journal: Phase Transitions / Year: 1992Title: Two Forms of Pf1 Inovirus: X-Ray Diffraction Studies on a Structural Phase Transition and a Calculated Libration Normal Mode of the Asymmetric Unit Authors: Marvin, D.A. / Nave, C. / Bansal, M. / Hale, R.D. / Salje, E.K.H. #3: Journal: Int.J.Biol.Macromol. / Year: 1990Title: Model-Building Studies of Inovirus: Genetic Variations on a Geometric Theme Authors: Marvin, D.A. #4: Journal: Int.J.Biol.Macromol. / Year: 1990Title: Erratum. Model-Building Studies of Inovirus: Genetic Variations on a Geometric Theme Authors: Marvin, D.A. #5: Journal: Int.J.Biol.Macromol. / Year: 1989Title: Dynamics of Telescoping Inovirus: A Mechanism for Assembly at Membrane Adhesions Authors: Marvin, D.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ifp.cif.gz | 17.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ifp.ent.gz | 10.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1ifp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/if/1ifpftp://data.pdbj.org/pub/pdb/validation_reports/if/1ifp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ifnS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 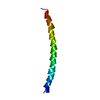
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 35
| ||||||||
| 2 |
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
| ||||||||
| Symmetry | Helical symmetry: (Circular symmetry: 1 / Dyad axis: no / N subunits divisor: 1 / Num. of operations: 35 / Rise per n subunits: 2.9 Å / Rotation per n subunits: 65.667 °) |
-Components
| #1: Protein/peptide | Mass: 4632.466 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas phage Pf3 (virus) / Genus: Inovirus / Strain: NEW YORK / Description: FILAMENTOUS BACTERIOPHAGE / Production host:   Pseudomonas aeruginosa (bacteria) / References: UniProt: P03623 Pseudomonas aeruginosa (bacteria) / References: UniProt: P03623 |
|---|
-Experimental details
-Experiment
| Experiment | Method: FIBER DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | pH: 7 / Details: pH 7.0 |
|---|---|
| Crystal grow | *PLUS Method: vapor diffusion |
| Components of the solutions | *PLUS Conc.: 30 mg/ml / Common name: protein |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.488 / Beamline: PX7.2 / Wavelength: 1.488 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Dec 1, 1995 / Details: MIRRORS |
| Radiation | Monochromator: GE(111) / Monochromatic (M) / Laue (L): M |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→60 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1IFN Resolution: 3.1→12 Å / Rfactor Rfree error: 0.01 / Cross valid method: A POSTERIORI Details: MODEL REFINED WITH A VERSION OF X-PLOR 3.1 MODIFIED FOR USE WITH FIBRE DIFFRACTION DATA BY WANG & STUBBS (1993) ACTA CRYST. A49, 504-513.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 19 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.1→12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: STRICT NCS WAS IMPOSED ON THE MODEL THROUGHOUT THE REFINEMENT. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.1→3.43 Å / Total num. of bins used: 4 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
