 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h9d | ||||||
|---|---|---|---|---|---|---|---|
| Title | Aml1/cbf-beta/dna complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSCRIPTION FACTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of connective tissue replacement / peripheral nervous system neuron development / positive regulation of granulocyte differentiation / RUNX3 regulates RUNX1-mediated transcription / RUNX1 regulates transcription of genes involved in BCR signaling / SLC-mediated transport of organic cations / RUNX1 regulates transcription of genes involved in interleukin signaling / core-binding factor complex / RUNX1 regulates expression of components of tight junctions / RUNX2 regulates bone development ...regulation of connective tissue replacement / peripheral nervous system neuron development / positive regulation of granulocyte differentiation / RUNX3 regulates RUNX1-mediated transcription / RUNX1 regulates transcription of genes involved in BCR signaling / SLC-mediated transport of organic cations / RUNX1 regulates transcription of genes involved in interleukin signaling / core-binding factor complex / RUNX1 regulates expression of components of tight junctions / RUNX2 regulates bone development / positive regulation of CD8-positive, alpha-beta T cell differentiation / RUNX2 regulates chondrocyte maturation / regulation of cardiac muscle cell proliferation / myeloid leukocyte differentiation / cardiac muscle tissue regeneration / negative regulation of CD4-positive, alpha-beta T cell differentiation / negative regulation of granulocyte differentiation / RUNX1 and FOXP3 control the development of regulatory T lymphocytes (Tregs) / positive regulation of extracellular matrix organization / RUNX2 regulates genes involved in cell migration / Transcriptional regulation by RUNX2 / myeloid cell differentiation / regulation of plasminogen activation / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / RUNX2 regulates genes involved in differentiation of myeloid cells / RUNX3 Regulates Immune Response and Cell Migration / hematopoietic stem cell proliferation / RUNX1 regulates transcription of genes involved in differentiation of myeloid cells / Regulation of RUNX1 Expression and Activity / RUNX1 regulates transcription of genes involved in WNT signaling / RUNX1 regulates estrogen receptor mediated transcription / RUNX2 regulates osteoblast differentiation / hemopoiesis / chondrocyte differentiation / positive regulation of collagen biosynthetic process / regulation of cell differentiation / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / RUNX3 regulates p14-ARF / ossification / positive regulation of interleukin-2 production / transcription corepressor binding / RNA polymerase II transcription regulatory region sequence-specific DNA binding / RUNX1 regulates genes involved in megakaryocyte differentiation and platelet function / Regulation of RUNX3 expression and activity / Pre-NOTCH Transcription and Translation / transcription coactivator binding / Transcriptional regulation of granulopoiesis / positive regulation of angiogenesis / neuron differentiation / protein polyubiquitination / SARS-CoV-1 activates/modulates innate immune responses / Regulation of RUNX2 expression and activity / transcription by RNA polymerase II / RUNX1 regulates transcription of genes involved in differentiation of HSCs / DNA-binding transcription activator activity, RNA polymerase II-specific / Estrogen-dependent gene expression / DNA-binding transcription factor binding / sequence-specific DNA binding / DNA-binding transcription factor activity, RNA polymerase II-specific / transcription coactivator activity / transcription cis-regulatory region binding / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / protein heterodimerization activity / calcium ion binding / regulation of transcription by RNA polymerase II / positive regulation of DNA-templated transcription / chromatin / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / protein homodimerization activity / DNA binding / nucleoplasm / ATP binding / membrane / nucleus Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Bravo, J. / Warren, A.J. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2001 Title: The Leukemia-Associated Aml1 (Runx1)-Cbfbeta Complex Functions as a DNA-Induced Molecular Clamp Authors: Bravo, J. / Li, Z. / Speck, N.A. / Warren, A.J. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h9d.cif.gz | 136.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h9d.ent.gz | 103.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1h9d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h9/1h9dftp://data.pdbj.org/pub/pdb/validation_reports/h9/1h9d | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1e50S S: Starting model for refinement |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 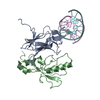
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14909.090 Da / Num. of mol.: 2 / Fragment: RUNT DOMAIN RESIDUES 50-183 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Gene: AML1 / Plasmid: PRSET / Production host:  #2: Protein | Mass: 15815.746 Da / Num. of mol.: 2 / Fragment: HETERODIMERISATION DOMAIN RESIDUES 2-135 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Gene: CBFB / Plasmid: PRSET / Production host: #3: DNA chain | Mass: 3107.025 Da / Num. of mol.: 2 / Source method: obtained synthetically #4: DNA chain | Mass: 2982.984 Da / Num. of mol.: 2 / Source method: obtained synthetically #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 56 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 56.8 % / Description: ON HOLD | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 / Details: pH 6.50 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 23 ℃ / pH: 6 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-3 / Wavelength: 0.9366 / Beamline: ID14-3 / Wavelength: 0.9366 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Oct 15, 2000 / Details: MIRRORS |
| Radiation | Monochromator: DIAMOND (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9366 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→50 Å / Num. obs: 27926 / % possible obs: 99.1 % / Redundancy: 6 % / Biso Wilson estimate: 60.1 Å2 / Rsym value: 0.126 / Net I/σ(I): 19.4 |
| Reflection shell | Resolution: 2.6→2.64 Å / Mean I/σ(I) obs: 2.2 / Rsym value: 0.38 / % possible all: 98.8 |
| Reflection | *PLUS Num. measured all: 193723 / Rmerge(I) obs: 0.126 |
| Reflection shell | *PLUS % possible obs: 98.8 % / Rmerge(I) obs: 0.38 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E50 Resolution: 2.6→48.02 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1104447.41 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 44.3898 Å2 / ksol: 0.380588 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 48.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→48.02 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.76 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.36 |
