SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" AND "BA" IN EACH CHAIN ON SHEET ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" AND "BA" IN EACH CHAIN ON SHEET RECORDS BELOW ARE ACTUALLY A 9-STRANDED BARREL THIS IS REPRESENTED BY A 10-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Mass: 18.015 Da / Num. of mol.: 658 / Source method: isolated from a natural source / Formula: H2O
-
Details
Compound details
THE ASYMMETRIC UNIT CONTAINS A DIMER WHICH BY MEANS OF A CRYSTALLOGRAPHIC TWO-FOLD SYMMETRY ...THE ASYMMETRIC UNIT CONTAINS A DIMER WHICH BY MEANS OF A CRYSTALLOGRAPHIC TWO-FOLD SYMMETRY OPERATOR FORMS THE FUNCTIONAL TETRAMERIC ENZYME
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 3
-
Sample preparation
Crystal
Density Matthews: 2.71 Å3/Da / Density % sol: 54.7 % Description: WAVELENGTHS QUOTED ARE FOR MAD PEAK, MAD INFLECTION POINT, MAD REMOTE 1, MAD REMOTE2, INHOUSE REFINEMENT, ID14-EH2 HIGH RES REFINEMENT DATA SETS RESPECTIVELY EXPERIMENTAL STATISTICS ...Description: WAVELENGTHS QUOTED ARE FOR MAD PEAK, MAD INFLECTION POINT, MAD REMOTE 1, MAD REMOTE2, INHOUSE REFINEMENT, ID14-EH2 HIGH RES REFINEMENT DATA SETS RESPECTIVELY EXPERIMENTAL STATISTICS QUOTED FOR COMBINED INHOUSE AND HIGH RES DATA SET USED FOR REFINEMENT
Resolution: 1.45→29.8 Å / Num. obs: 1086028 / % possible obs: 98.4 % / Redundancy: 3 % / Rmerge(I) obs: 0.064 / Net I/σ(I): 15
Reflection shell
Resolution: 1.45→1.48 Å / Redundancy: 3 % / Rmerge(I) obs: 0.292 / Mean I/σ(I) obs: 1.5 / % possible all: 97.9
Reflection
*PLUS
Num. obs: 131455 / Num. measured all: 1086028
-
Processing
Software
Name
Classification
DENZO
datareduction
MOSFLM
datareduction
SCALEPACK
datascaling
SCALA
datascaling
RSPS
phasing
MLPHARE
phasing
SHELXL-97
refinement
Refinement
Method to determine structure: DIRECT METHODS / Resolution: 1.45→10 Å / Num. parameters: 47082 / Num. restraintsaints: 56585 / Cross valid method: FREE R-VALUE / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ARP/WARP WITH REFMAC USED FIRST FOLLOWED BY CNS THE FOLLOWING RESIDUES HAVE MISSING ATOMS DUE TO DISORDER: SER A 151 LYS A 185 LYS A 188 ARG A 257 SER A 285 GLU B 150 SER B 151 LYS B 185 LYS B 188 GLU B 216
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.1733
6592
5.3 %
RANDOM
all
0.1268
124490
-
-
obs
0.131
-
93.4 %
-
Solvent computation
Solvent model: MOEWS & KRETSINGER
Refine analyze
Num. disordered residues: 15 / Occupancy sum hydrogen: 4167 / Occupancy sum non hydrogen: 4926.25
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj
 Assembly
Assembly

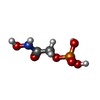




 Mass: 171.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6NO6P
Mass: 171.046 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6NO6P Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6O2 Sample preparation
Sample preparation / Beamline: BM14 / Wavelength: 1.282,1.283,1.127,1.030, 1.5418,0.933
/ Beamline: BM14 / Wavelength: 1.282,1.283,1.127,1.030, 1.5418,0.933 Processing
Processing