 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g6n | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 2.1 ANGSTROM STRUCTURE OF CAP-CAMP | |||||||||
 Components Components | CATABOLITE GENE ACTIVATOR PROTEIN | |||||||||
 Keywords Keywords | DNA BINDING PROTEIN / Catabolite activator protein (CAP) / cAMP receptor protein (CRP) / transcription / allostery / cyclic AMP / cAMP | |||||||||
| Function / homology |  Function and homology information Function and homology informationcarbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / DNA-templated transcription ...carbon catabolite repression of transcription / DNA binding, bending / minor groove of adenine-thymine-rich DNA binding / cAMP binding / protein-DNA complex / sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / DNA-templated transcription / identical protein binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | |||||||||
 Authors Authors | Passner, J.M. / Schultz, S.C. / Steitz, T.A. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: Modeling the cAMP-induced allosteric transition using the crystal structure of CAP-cAMP at 2.1 A resolution. Authors: Passner, J.M. / Schultz, S.C. / Steitz, T.A. #1: Journal: J.MOL.BIOL. / Year: 1987Title: Structure of a complex of catabolite gene activator protein and cyclic AMP at 2.5 A resolution Authors: Weber, I.T. / Steitz, T.A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g6n.cif.gz | 93.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g6n.ent.gz | 70.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1g6n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g6/1g6nftp://data.pdbj.org/pub/pdb/validation_reports/g6/1g6n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3gap S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23672.439 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | 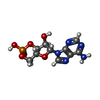  Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 115 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.64 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: microdialysis / pH: 7.5 Details: 0.1mM cAMP, 0.2M sodium chloride, 5mM Tris, O.1mM EDTA, 2mM DTT, .02% sodium azide, pH 7.5, MICRODIALYSIS, temperature 298.0K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 253 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 Å |
| Detector | Type: UCSD MARK III / Detector: AREA DETECTOR / Date: Mar 1, 1990 |
| Radiation | Monochromator: GRAPHITE + NICKLE FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.97→25 Å / Num. obs: 31208 / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3 % / Rmerge(I) obs: 0.0525 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 1.97→2.12 Å / Redundancy: 1.9 % / Mean I/σ(I) obs: 1.37 |
| Reflection | *PLUS Num. measured all: 90177 |
| Reflection shell | *PLUS Num. unique obs: 4506 / Num. measured obs: 8697 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3GAP 3gap Resolution: 2.1→12 Å / Isotropic thermal model: Isotropic / σ(F): 2 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→12 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.843 / Classification: refinement | ||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_bond_d / Dev ideal: 0.008 |
