 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1g3l: THE STRUCTURAL BASIS OF THE CATALYTIC MECHANISM AND REGULATION OF... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1g3l | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE STRUCTURAL BASIS OF THE CATALYTIC MECHANISM AND REGULATION OF GLUCOSE-1-PHOSPHATE THYMIDYLYLTRANSFERASE (RMLA). TDP-L-RHAMNOSE COMPLEX. | ||||||
 Components Components | GLUCOSE-1-PHOSPHATE THYMIDYLYLTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / L-RHAMNOSE / NUCLEOTIDYLTRANSFERASE / PYROPHOSPHORYLASE / THYMIDYLYLTRANSFERASE / ALLOSTERY | ||||||
| Function / homology |  Function and homology information Function and homology informationglucose-1-phosphate thymidylyltransferase / glucose-1-phosphate thymidylyltransferase activity / dTDP-rhamnose biosynthetic process / lipopolysaccharide core region biosynthetic process / polysaccharide biosynthetic process / nucleotide binding / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.7 Å X-RAY DIFFRACTION / Resolution: 2.7 Å | ||||||
 Authors Authors | Blankenfeldt, W. / Asuncion, M. / Lam, J.S. / Naismith, J.H. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 2000 Title: The structural basis of the catalytic mechanism and regulation of glucose-1-phosphate thymidylyltransferase (RmlA). Authors: Blankenfeldt, W. / Asuncion, M. / Lam, J.S. / Naismith, J.H. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2000Title: The Purification, Crystallisation and Preliminary Structural Characterisation of Glucose-1-Phosphate Thymidylyltransferase (Rmla), the First Enzyme of the Dtdp-L-Rhamnose Synthesis Pathway ...Title: The Purification, Crystallisation and Preliminary Structural Characterisation of Glucose-1-Phosphate Thymidylyltransferase (Rmla), the First Enzyme of the Dtdp-L-Rhamnose Synthesis Pathway from Pseudomonas Aeruginosa Authors: Blankenfeldt, W. / Giraud, M.F. / Leonard, G. / Rahim, R. / Creuzenet, C. / Lam, J.S. / Naismith, J.H. #2: Journal: J.Biol.Chem. / Year: 1965Title: The Nucleotide Specificity and Feedback Control of Thymidine Diphosphate D-Glucose Pyrophosphorylase. Authors: Melo, A. / Glaser, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1g3l.cif.gz | 235.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1g3l.ent.gz | 192.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1g3l.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/g3/1g3lftp://data.pdbj.org/pub/pdb/validation_reports/g3/1g3l | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1fxoC  1fzwC  1g0rC  1g1lSC  1g23C  1g2vC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer consisting of two dimers. |
-Components
| #1: Protein | Mass: 32488.844 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Plasmid: PET23A(+) / Production host: References: UniProt: Q9HU22, glucose-1-phosphate thymidylyltransferase #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-TRH / 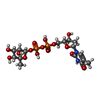  Mass: 548.330 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C16H26N2O15P2 Mass: 548.330 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C16H26N2O15P2 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 45.2 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 4 Details: 10 % (w/v) PEG 6000, 0.2 M Li-sulfate, 0.1 M Na-citrate pH 4.0; protein incubated with 10 mM dTDP-L-rhamnose, VAPOR DIFFUSION, SITTING DROP, temperature 292K | ||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Mar 1, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→49.9 Å / Num. obs: 36311 / % possible obs: 94.2 % / Observed criterion σ(I): 0 / Redundancy: 7.2 % / Biso Wilson estimate: 37.11 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 6.8 |
| Reflection shell | Resolution: 2.8→2.84 Å / Redundancy: 5.7 % / Rmerge(I) obs: 0.266 / Mean I/σ(I) obs: 2.9 / % possible all: 70.8 |
| Reflection | *PLUS Num. measured all: 260832 |
| Reflection shell | *PLUS % possible obs: 70.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: 1G1L Resolution: 2.7→100 Å / SU ML: 0.41 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.47
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.78 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.333 / Rfactor Rwork: 0.255 |
