 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1eu1: THE CRYSTAL STRUCTURE OF RHODOBACTER SPHAEROIDES DIMETHYLSULFOXID... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eu1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THE CRYSTAL STRUCTURE OF RHODOBACTER SPHAEROIDES DIMETHYLSULFOXIDE REDUCTASE REVEALS TWO DISTINCT MOLYBDENUM COORDINATION ENVIRONMENTS. | |||||||||
 Components Components | DIMETHYL SULFOXIDE REDUCTASE | |||||||||
 Keywords Keywords | OXIDOREDUCTASE / Molybdenum / Molybdenum Cofactor / DMSO / Reductase / Molybdopterin / MGD | |||||||||
| Function / homology |  Function and homology information Function and homology informationrespiratory dimethylsulfoxide reductase / trimethylamine-N-oxide reductase / trimethylamine-N-oxide reductase (cytochrome c) activity / molybdenum ion binding / molybdopterin cofactor binding / anaerobic respiration / outer membrane-bounded periplasmic space / electron transfer activity Similarity search - Function | |||||||||
| Biological species |  Rhodobacter sphaeroides (bacteria) Rhodobacter sphaeroides (bacteria) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.3 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.3 Å | |||||||||
 Authors Authors | Li, H.K. / Temple, K. / Rajagopalan, K.V. / Schindelin, H. | |||||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2000 Title: The 1.3 A Crystal Structure of Rhodobacter sphaeroides Dimethylsulfoxide Reductase Reveals Two Distinct Molybdenum Coordination Environments Authors: Li, H.K. / Temple, K. / Rajagopalan, K.V. / Schindelin, H. #1: Journal: Science / Year: 1996Title: Crystal Structure of DMSO Reductase: Redox-linked Changes in Molybdopterin Coordination | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eu1.cif.gz | 195.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eu1.ent.gz | 150.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1eu1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eu/1eu1ftp://data.pdbj.org/pub/pdb/validation_reports/eu/1eu1 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Sugars , 2 types, 4 molecules A
| #1: Protein | Mass: 85022.367 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Rhodobacter sphaeroides (bacteria) / Production host: Bacteria (eubacteria)References: UniProt: Q57366, Oxidoreductases; Acting on a sulfur group of donors; With unknown physiological acceptors |
|---|---|
| #2: Sugar |  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
-Non-polymers , 7 types, 1047 molecules 





| #3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #4: Chemical | ChemComp-CD /  Mass: 112.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cd Mass: 112.411 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cd | ||||||||
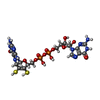
| #5: Chemical |  Mass: 740.557 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H26N10O13P2S2 Mass: 740.557 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C20H26N10O13P2S2#6: Chemical | ChemComp-6MO / |  Mass: 95.940 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mo Mass: 95.940 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mo#7: Chemical |  Mass: 15.999 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: O Mass: 15.999 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: O#8: Chemical | ChemComp-EPE / |  Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#9: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1039 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.55 Å3/Da / Density % sol: 51.68 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 7 Details: Hepes, Cadmium Chloride, Ammonium Sulfate, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 22.0K | ||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 52 % | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X26C / Wavelength: 1.1 / Beamline: X26C / Wavelength: 1.1 |
| Detector | Type: ADSC / Detector: CCD / Date: Feb 4, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→50 Å / Num. all: 213176 / Num. obs: 191461 / % possible obs: 89.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.6 % / Biso Wilson estimate: 11.5 Å2 / Rmerge(I) obs: 0.051 / Net I/σ(I): 19.9 |
| Reflection shell | Resolution: 1.3→1.35 Å / Rmerge(I) obs: 0.211 / Num. unique all: 12690 / % possible all: 60.2 |
| Reflection | *PLUS Highest resolution: 1.3 Å |
| Reflection shell | *PLUS Highest resolution: 1.3 Å / % possible obs: 60.2 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.3→10 Å / σ(F): 0 / σ(I): 0 Stereochemistry target values: Victor S. Lamzin, Zbigniew Dauter, and Keith S. Wilson Details: Maximum likelihood refinement including anisotropic temperature factors for all atoms.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→10 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.3 Å / σ(F): 0 / Rfactor obs: 0.121 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.3 Å / Lowest resolution: 1.36 Å / Rfactor Rfree: 0.187 / Rfactor obs: 0.161 |
