 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ene: 1A CRYSTAL STRUCTURES OF B-DNA REVEAL SEQUENCE-SPECIFIC BINDING A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ene | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | 1A CRYSTAL STRUCTURES OF B-DNA REVEAL SEQUENCE-SPECIFIC BINDING AND GROOVE-SPECIFIC BENDING OF DNA BY MAGNESIUM AND CALCIUM. | ||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / divalent cations / DNA sequence-specific binding / shelxdna / B-DNA | Function / homology | ETHANOL / DNA |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.985 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 0.985 Å  Authors AuthorsChiu, T.K. / Dickerson, R.E. |  CitationJournal: J.Mol.Biol. / Year: 2000 CitationJournal: J.Mol.Biol. / Year: 2000Title: 1 A crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium. Authors: Chiu, T.K. / Dickerson, R.E. #1: Journal: J.Mol.Biol. / Year: 1999Title: Absence of minor groove monovalent cations in the crosslinked dodecamer CGCGAATTCGCG Authors: Chiu, T.K. / Kaczor-Grzeskowiak, M. / Dickerson, R.E. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ene.cif.gz | 33.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ene.ent.gz | 22.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1ene.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/en/1eneftp://data.pdbj.org/pub/pdb/validation_reports/en/1ene | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1en3C  1en8C  1en9C C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |






-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 3045.992 Da / Num. of mol.: 1 / Source method: obtained synthetically | ||||
|---|---|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Ca#3: Chemical | 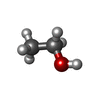  Mass: 46.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O Mass: 46.068 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C2H6O#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 89 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 36.8 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 275 K / Method: vapor diffusion, sitting drop Details: initial concentrations in droplet: 0.17 mM dna, 61.20 mM calcium acetate, 10-15% MPD, 30% final MPD concentration in reservoir. Solutions were unbuffered, VAPOR DIFFUSION, SITTING DROP, temperature 275.0K | |||||||||||||||||||||||||
| Components of the solutions |
| |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Details: sitting drop vapor diffusion in nine-well plates | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X8C / Wavelength: 0.95 / Beamline: X8C / Wavelength: 0.95 |
| Detector | Type: MARRESEARCH / Detector: AREA DETECTOR / Date: Nov 19, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 |
| Reflection | Resolution: 0.985→8 Å / Num. all: 13423 / Num. obs: 13423 / % possible obs: 95.5 % / Observed criterion σ(I): -1 / Redundancy: 4.65 % / Biso Wilson estimate: 4.41 Å2 / Rmerge(I) obs: 0.032 / Net I/σ(I): 30.7 |
| Reflection shell | Resolution: 0.985→1.016 Å / Rmerge(I) obs: 0.09 / Mean I/σ(I) obs: 9.1 / Num. unique all: 1279 / % possible all: 91.5 |
| Reflection | *PLUS Num. measured all: 62360 |
| Reflection shell | *PLUS % possible obs: 91.5 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: BDJ019 Resolution: 0.985→8 Å / Num. parameters: 16552 / Num. restraintsaints: 4814 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Parkinson et al. Details: REFINEMENT STARTED IN X-PLOR 3.843 WITH IDEAL B-DNA FITTED ONTO BDJ019. AFTER ALL DATA HAS BEEN ADDED AND REFINED BY SIMULATED ANNEALING IN X-PLOR 3.843, REFINEMENT CONTINUED BY CONJUGATE ...Details: REFINEMENT STARTED IN X-PLOR 3.843 WITH IDEAL B-DNA FITTED ONTO BDJ019. AFTER ALL DATA HAS BEEN ADDED AND REFINED BY SIMULATED ANNEALING IN X-PLOR 3.843, REFINEMENT CONTINUED BY CONJUGATE GRADIENT LEAST-SQUARES IN SHELXL-97. THE TOP 50 MOST DISAGREEABLE REFLECTIONS WERE REJECTED TOWARDS THE LATTER STAGES OF REFINEMENT BUT THESE ARE STILL INCLUDED IN THE RELEASED DATA.
| |||||||||||||||||||||||||
| Solvent computation | Solvent model: SHELX swat option | |||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 13 / Occupancy sum hydrogen: 113 / Occupancy sum non hydrogen: 276.5 | |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 0.985→8 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| |||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 0.985 Å / Lowest resolution: 1.016 Å / Rfactor Rfree: 0.1629 / Rfactor Rwork: 0.139 |
