 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eax | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of MTSP1 (matriptase) | ||||||
 Components Components | SUPPRESSOR OF TUMORIGENICITY 14 | ||||||
 Keywords Keywords | HYDROLASE / SERINE PROTEINASE / MATRIX DEGRADATION | ||||||
| Function / homology |  Function and homology information Function and homology informationmatriptase / Formation of the cornified envelope / keratinocyte differentiation / protein catabolic process / serine-type peptidase activity / protein processing / basolateral plasma membrane / serine-type endopeptidase activity / external side of plasma membrane / proteolysis ...matriptase / Formation of the cornified envelope / keratinocyte differentiation / protein catabolic process / serine-type peptidase activity / protein processing / basolateral plasma membrane / serine-type endopeptidase activity / external side of plasma membrane / proteolysis / : / plasma membrane Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.3 Å | ||||||
 Authors Authors | Friedrich, R. / Bode, W. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Catalytic Domain Structures of Mt-Sp1/Matriptase, a Matrix-Degrading Transmembrane Serine Proteinase. Authors: Friedrich, R. / Fuentes-Prior, P. / Ong, E. / Coombs, G. / Hunter, M. / Oehler, R. / Pierson, D. / Gonzalez, R. / Huber, R. / Bode, W. / Madison, E.L. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eax.cif.gz | 69.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eax.ent.gz | 51.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1eax.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ea/1eaxftp://data.pdbj.org/pub/pdb/validation_reports/ea/1eax | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1eawC  1ekbS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 26463.756 Da / Num. of mol.: 1 / Fragment: CATALYTIC RESIDUES 615-855 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  References: UniProt: Q9Y5Y6, Hydrolases; Acting on peptide bonds (peptidases); Serine endopeptidases |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-BEN / 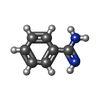  Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 Mass: 120.152 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8N2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 381 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 381 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 / Details: pH 8.00 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.3→18 Å / Num. obs: 58805 / % possible obs: 96.2 % / Observed criterion σ(I): 2 / Redundancy: 3.3 % / Biso Wilson estimate: 12.1 Å2 / Rmerge(I) obs: 0.086 / Net I/σ(I): 4 |
| Reflection shell | Resolution: 1.3→1.36 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.294 / Mean I/σ(I) obs: 1.3 / % possible all: 93.5 |
| Reflection | *PLUS Highest resolution: 1.3 Å / Lowest resolution: 18 Å |
| Reflection shell | *PLUS % possible obs: 93.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1EKB Resolution: 1.3→17.75 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 1648979.68 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 65 Å2 / ksol: 0.388634 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.3→17.75 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.3→1.38 Å / Rfactor Rfree error: 0.012 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 18 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.25 |
