 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1e8c: Structure of MurE the UDP-N-acetylmuramyl tripeptide synthetase f... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1e8c | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of MurE the UDP-N-acetylmuramyl tripeptide synthetase from E. coli | |||||||||
 Components Components | UDP-N-ACETYLMURAMOYLALANYL-D-GLUTAMATE--2,6-DIAMINOPIMELATE LIGASE | |||||||||
 Keywords Keywords | LIGASE / PEPTIDOGLYCAN BIOSYNTHESIS | |||||||||
| Function / homology |  Function and homology information Function and homology informationUDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase / UDP-N-acetylmuramoylalanyl-D-glutamate-2,6-diaminopimelate ligase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / cell division / magnesium ion binding / ATP binding / cytosol Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2 Å | |||||||||
 Authors Authors | Gordon, E.J. / Chantala, L. / Dideberg, O. | |||||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Crystal Structure of Udp-N-Acetylmuramoyl-L-Alanyl-D-Glutamate: Meso-Diaminopimelate Ligase from Escherichia Coli Authors: Gordon, E.J. / Flouret, B. / Chantalat, L. / Van Heijenoort, J. / Mengin-Lecreulx, D. / Dideberg, O. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1e8c.cif.gz | 213.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1e8c.ent.gz | 170.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1e8c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e8/1e8cftp://data.pdbj.org/pub/pdb/validation_reports/e8/1e8c | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|

-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.042, -0.999, -0.012), Vector: |
-Components
| #1: Protein | Mass: 54401.535 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: UDP-N-ACETYLMURAMYL-TRIPEPTIDE BOUND IN ACTIVE SITE Source: (gene. exp.) References: UniProt: P22188, UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase #2: Chemical | ChemComp-CL / |   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#3: Chemical | 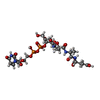  Mass: 879.608 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H43N5O23P2 Mass: 879.608 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C28H43N5O23P2#4: Chemical | 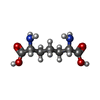  Type: L-peptide linking / Mass: 190.197 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H14N2O4 Type: L-peptide linking / Mass: 190.197 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H14N2O4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 393 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 393 / Source method: isolated from a natural source / Formula: H2OCompound details | RESIDUES AFTER 494 BELONG TO LINKER AND HIS TAG USED TO EXPRESS AND PURIFY THE PROTEIN | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.8 Å3/Da / Density % sol: 47 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 7.5 Details: HANGING DROP EXPERIMENT RESERVOIR: 13% PEG MME 5K, 0.5M LICL, 10% ISOPROPANOL, 0.1M HEPES PH 7.5, 5MM DTT, 1MM UDP-TRIPEPTIDE DROP: 2UL PROTEIN SOLUTION (MURE @ 10MGML-1 IN 20MM HEPES PH 7. ...Details: HANGING DROP EXPERIMENT RESERVOIR: 13% PEG MME 5K, 0.5M LICL, 10% ISOPROPANOL, 0.1M HEPES PH 7.5, 5MM DTT, 1MM UDP-TRIPEPTIDE DROP: 2UL PROTEIN SOLUTION (MURE @ 10MGML-1 IN 20MM HEPES PH 7.5, 200MM NACL, 5MM DTT) WITH 2UL RESERVOIR SOLUTION. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.9791,0.9790,0.8550,0.93 / Beamline: BM14 / Wavelength: 0.9791,0.9790,0.8550,0.93 | |||||||||||||||
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Nov 15, 1999 | |||||||||||||||
| Radiation | Monochromator: GE / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2→46.7 Å / Num. obs: 323548 / % possible obs: 98.3 % / Redundancy: 4.5 % / Biso Wilson estimate: 19.3 Å2 / Rsym value: 0.066 | |||||||||||||||
| Reflection shell | Resolution: 2→2.13 Å / Redundancy: 3.9 % / Rsym value: 0.22 / % possible all: 91.2 | |||||||||||||||
| Reflection | *PLUS Num. obs: 72674 / Num. measured all: 323548 / Rmerge(I) obs: 0.066 | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 91.2 % / Rmerge(I) obs: 0.22 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2→46.73 Å / Rfactor Rfree error: 0.003 / Data cutoff high absF: 3518491.82 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 38.2391 Å2 / ksol: 0.318364 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.4 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→46.73 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.008 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.25 |
