 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1e3j: Ketose reductase (sorbitol dehydrogenase) from silverleaf whitefly -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1e3j | ||||||
|---|---|---|---|---|---|---|---|
| Title | Ketose reductase (sorbitol dehydrogenase) from silverleaf whitefly | ||||||
 Components Components | NADP(H)-DEPENDENT KETOSE REDUCTASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FRUCTOSE REDUCTION | ||||||
| Function / homology |  Function and homology information Function and homology informationD-sorbitol catabolic process / L-iditol 2-dehydrogenase (NAD+) activity / zinc ion binding Similarity search - Function | ||||||
| Biological species |  BEMISIA ARGENTIFOLII (silverleaf whitefly) BEMISIA ARGENTIFOLII (silverleaf whitefly) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.3 Å | ||||||
 Authors Authors | Banfield, M.J. / Salvucci, M.E. / Baker, E.N. / Smith, C.A. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2001 Title: Crystal Structure of Nadp(H)-Dependent Ketose Reductase from Besimia Argentifolii at 2.3 Angstrom Resolution Authors: Banfield, M.J. / Salvucci, M.E. / Baker, E.N. / Smith, C.A. #1: Journal: J.Mol.Biol. / Year: 1998Title: Nadp-Dependent Bacterial Alcohol Dehydrogenases: Crystal Structure, Co-Factor-Binding and Co-Factor Specificity of the Adhs of Clostridium Beijerinckii and Thermoanaerobacter Brockii Authors: Korkhin, Y. / Kalb, A.J. / Peretz, M. / Bogin, O. / Burstein, Y. / Frolow, F. #2: Journal: J.Mol.Biol. / Year: 1976 Title: Nadp-Dependent Bacterial Alcohol Dehydrogenases: Crystal Structure, Co-Factor-Binding and Co-Factor Specificity of the Adhs of Clostridium Beijerinckii and Thermoanaerobacter Brockii Authors: Eklund, H. / Nordstrom, B. / Zeppezauer, E. / Soderlund, G. / Ohlsson, I. / Boiwe, T. / Soderberg, B.O. / Tapia, O. / Branden, C.I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1e3j.cif.gz | 78.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1e3j.ent.gz | 59.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1e3j.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e3/1e3jftp://data.pdbj.org/pub/pdb/validation_reports/e3/1e3j | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 38213.203 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) BEMISIA ARGENTIFOLII (silverleaf whitefly)Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-PO4 / |   Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4#4: Chemical | ChemComp-BO3 / | 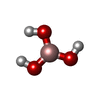  Mass: 61.833 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BH3O3 Mass: 61.833 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: BH3O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 47 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 47 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.06 Å3/Da / Density % sol: 53.3 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.75 Details: 1.4M AMMONIUM SUPLHATE, 6% (V/V) GLYCERO 100MM BORIC ACID, PH8.75 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 18 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-2 / Wavelength: 0.9998, 1.1807, 1.2825, 1.2829 / Beamline: BL9-2 / Wavelength: 0.9998, 1.1807, 1.2825, 1.2829 | |||||||||||||||
| Detector | Type: ADSC QUANTUM / Detector: CCD / Date: Feb 15, 2000 / Details: MIRRORS | |||||||||||||||
| Radiation | Monochromator: MIRRORS / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2.3→22 Å / Num. obs: 21214 / % possible obs: 97.5 % / Redundancy: 13.3 % / Biso Wilson estimate: 41.1 Å2 / Rmerge(I) obs: 0.049 / Net I/σ(I): 44.9 | |||||||||||||||
| Reflection shell | Resolution: 2.3→2.4 Å / Redundancy: 11.3 % / Rmerge(I) obs: 0.205 / Mean I/σ(I) obs: 7.9 / % possible all: 95.5 | |||||||||||||||
| Reflection | *PLUS Lowest resolution: 22 Å | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 95.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.3→20 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 2766186.47 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE C-TERMINAL RESIDUE WAS NOT SEEN I THE DENSITY MAPS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 39.7918 Å2 / ksol: 0.314832 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 49.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.44 Å / Rfactor Rfree error: 0.027 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor Rfree: 0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Lowest resolution: 2.38 Å / Rfactor Rfree: 0.345 / Rfactor obs: 0.27 |
