 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cx4: CRYSTAL STRUCTURE OF A DELETION MUTANT OF THE TYPE II BETA REGULA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cx4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF A DELETION MUTANT OF THE TYPE II BETA REGULATORY SUBUNIT OF CAMP-DEPENDENT PROTEIN KINASE | ||||||
 Components Components | CAMP-DEPENDENT PROTEIN KINASE REGULATORY SUBUNIT TYPE II BETA | ||||||
 Keywords Keywords | SIGNALING PROTEIN / BETA BARREL / CAMP-DEPENDENT PROTEIN KINASE / CAMP-BINDING / REGULATORY SUBUNIT | ||||||
| Function / homology |  Function and homology information Function and homology informationPKA activation in glucagon signalling / CREB1 phosphorylation through the activation of Adenylate Cyclase / DARPP-32 events / GPER1 signaling / Factors involved in megakaryocyte development and platelet production / Loss of Nlp from mitotic centrosomes / Recruitment of mitotic centrosome proteins and complexes / Loss of proteins required for interphase microtubule organization from the centrosome / Recruitment of NuMA to mitotic centrosomes / Anchoring of the basal body to the plasma membrane ...PKA activation in glucagon signalling / CREB1 phosphorylation through the activation of Adenylate Cyclase / DARPP-32 events / GPER1 signaling / Factors involved in megakaryocyte development and platelet production / Loss of Nlp from mitotic centrosomes / Recruitment of mitotic centrosome proteins and complexes / Loss of proteins required for interphase microtubule organization from the centrosome / Recruitment of NuMA to mitotic centrosomes / Anchoring of the basal body to the plasma membrane / AURKA Activation by TPX2 / Regulation of PLK1 Activity at G2/M Transition / PKA activation / Hedgehog 'off' state / cAMP-dependent protein kinase regulator activity / response to antipsychotic drug / High laminar flow shear stress activates signaling by PIEZO1 and PECAM1:CDH5:KDR in endothelial cells / Vasopressin regulates renal water homeostasis via Aquaporins / cAMP-dependent protein kinase inhibitor activity / cAMP-dependent protein kinase complex / ciliary base / protein kinase A catalytic subunit binding / cAMP binding / negative regulation of cAMP/PKA signal transduction / fatty acid metabolic process / learning / dendritic shaft / modulation of chemical synaptic transmission / adenylate cyclase-activating G protein-coupled receptor signaling pathway / dendritic spine / postsynapse / protein domain specific binding / neuronal cell body / centrosome / ubiquitin protein ligase binding / dendrite / protein kinase binding / perinuclear region of cytoplasm / glutamatergic synapse / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.45 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.45 Å | ||||||
 Authors Authors | Diller, T.C. / Xuong, N.H. / Taylor, S.S. | ||||||
 Citation Citation | Journal: Structure / Year: 2001 Title: Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Authors: Diller, T.C. / Madhusudan / Xuong, N.H. / Taylor, S.S. #1: Journal: Protein Expr.Purif. / Year: 2000 Title: Type II beta regulatory subunit of cAMP-dependent protein kinase: purification strategies to optimize crystallization. Authors: Diller, T.C. / Xuong, N.H. / Taylor, S.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cx4.cif.gz | 73.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cx4.ent.gz | 54.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1cx4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cx/1cx4ftp://data.pdbj.org/pub/pdb/validation_reports/cx/1cx4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34157.719 Da / Num. of mol.: 1 / Fragment: CAMP BINDING DOMAINS Source method: isolated from a genetically manipulated source Source: (gene. exp.)  | ||
|---|---|---|---|
| #2: Chemical | 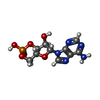  Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P Mass: 329.206 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H12N5O6P#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 192 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.38 Å3/Da / Density % sol: 70.8 % Description: PROTEIN RESIDUES PRECEDING ARG 130 AND POSTERIOR TO VAL 413 IN THE AMINO ACID SEQUENCE ARE NOT INCLUDED IN THIS MODEL. ADDITIONALLY, LOOP REGION CONSISTING OF RESIDUES 326 LYS THROUGH ...Description: PROTEIN RESIDUES PRECEDING ARG 130 AND POSTERIOR TO VAL 413 IN THE AMINO ACID SEQUENCE ARE NOT INCLUDED IN THIS MODEL. ADDITIONALLY, LOOP REGION CONSISTING OF RESIDUES 326 LYS THROUGH 333 GLU WERE NOT TRACED IN ELECTRON DENSITY MAPS: NO PEPTIDE BOND EXISTS BETWEEN ARG 325 AND ASN 334 IN THIS STRUCTURAL MODEL. | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.75 / Details: pH 7.75 | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: Diller, T.C., (2000) Protein Expression Purif., 20, 357. | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 300.3 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL7-1 / Wavelength: 1.08 / Beamline: BL7-1 / Wavelength: 1.08 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 6, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.08 Å / Relative weight: 1 |
| Reflection | Resolution: 2.45→25 Å / Num. obs: 21965 / % possible obs: 98.7 % / Observed criterion σ(I): 0 / Redundancy: 4.182 % / Biso Wilson estimate: 36.4 Å2 / Rmerge(I) obs: 0.052 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 2.45→2.56 Å / Redundancy: 4 % / Rmerge(I) obs: 0.325 / Mean I/σ(I) obs: 3.94 / % possible all: 97.9 |
| Reflection shell | *PLUS % possible obs: 97.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.45→23 Å / Rfactor Rfree error: 0.004 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER Details: INSUFFICIENT ELECTRON DENSITY EXISTED IN MAPS TO INCLUDE RESIDUE RANGES 112- 128, 326-333 AND 413-416.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 49.37 Å2 / ksol: 0.33 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.45→23 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.45→2.56 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 23 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
