 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cbx: CRYSTAL STRUCTURE OF THE COMPLEX BETWEEN CARBOXYPEPTIDASE A AND T... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cbx | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE COMPLEX BETWEEN CARBOXYPEPTIDASE A AND THE BIPRODUCT ANALOG INHIBITOR L-BENZYLSUCCINATE AT 2.0 ANGSTROMS RESOLUTION | ||||||
 Components Components | CARBOXYPEPTIDASE A | ||||||
 Keywords Keywords | HYDROLASE(C-TERMINAL PEPTIDASE) | ||||||
| Function / homology |  Function and homology information Function and homology informationcarboxypeptidase A / leukotriene metabolic process / metallocarboxypeptidase activity / proteolysis / : / zinc ion binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2 Å X-RAY DIFFRACTION / Resolution: 2 Å | ||||||
 Authors Authors | Mangani, S. / Carloni, P. / Orioli, P. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1992 Title: Crystal structure of the complex between carboxypeptidase A and the biproduct analog inhibitor L-benzylsuccinate at 2.0 A resolution. Authors: Mangani, S. / Carloni, P. / Orioli, P. #1: Journal: J.Mol.Biol. / Year: 1983Title: Refined Crystal Structure of Carboxypeptidase a at 1.54 Angstroms Resolution Authors: Rees, D.C. / Lewis, M. / Lipscomb, W.N. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE PRESENT STRUCTURE MAINTAINS ALL SECONDARY STRUCTURES (HELICES, SHEETS AND TURNS) OF THE ...SHEET THE PRESENT STRUCTURE MAINTAINS ALL SECONDARY STRUCTURES (HELICES, SHEETS AND TURNS) OF THE NATIVE CPA STRUCTURE OF PROTEIN DATA BANK ENTRY 5CPA. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cbx.cif.gz | 79.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cbx.ent.gz | 57.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1cbx.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cb/1cbxftp://data.pdbj.org/pub/pdb/validation_reports/cb/1cbx | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|


















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: SER 197 - TYR 198 OMEGA =342.52 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 2: TYR 198 - SER 199 OMEGA =145.05 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 3: PRO 205 - TYR 206 OMEGA = 11.68 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION 4: ARG 272 - ASP 273 OMEGA =357.86 PEPTIDE BOND DEVIATES SIGNIFICANTLY FROM TRANS CONFORMATION |
-Components
| #1: Protein | Mass: 34442.461 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
| #3: Chemical | ChemComp-BZS / 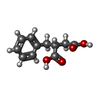  Mass: 208.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12O4 Mass: 208.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H12O4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 178 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THE SEQUENCE HAS BEEN TAKEN FROM PROTEIN DATA BANK ENTRY 5CPA. SEQUENCE ADVISORY NOTICE: DIFFERENCE ...THE SEQUENCE HAS BEEN TAKEN FROM PROTEIN DATA BANK ENTRY 5CPA. SEQUENCE ADVISORY NOTICE: DIFFERENCE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.12 Å3/Da / Density % sol: 41.85 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 7.5 / Method: microdialysis | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Radiation | Scattering type: x-ray |
|---|---|
| Radiation wavelength | Relative weight: 1 |
| Reflection | *PLUS Highest resolution: 2 Å / Num. obs: 22981 / Rmerge(I) obs: 0.055 |
- Processing
Processing
| Software | Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor obs: 0.166 / Highest resolution: 2 Å | ||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 2 Å
| ||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 10 Å / Num. reflection obs: 15835 / σ(I): 2 / Rfactor obs: 0.166 | ||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
