 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1a5i: CATALYTIC DOMAIN OF VAMPIRE BAT (DESMODUS ROTUNDUS) SALIVA PLASMI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a5i | ||||||
|---|---|---|---|---|---|---|---|
| Title | CATALYTIC DOMAIN OF VAMPIRE BAT (DESMODUS ROTUNDUS) SALIVA PLASMINOGEN ACTIVATOR IN COMPLEX WITH EGR-CMK (GLU-GLY-ARG CHLOROMETHYL KETONE) | ||||||
 Components Components | PLASMINOGEN ACTIVATOR | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / SERINE PROTEASE / FIBRINOLYTIC ENZYMES / PLASMINOGEN ACTIVATORS / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationt-plasminogen activator / smooth muscle cell migration / plasminogen activation / platelet-derived growth factor receptor signaling pathway / serine-type endopeptidase activity / calcium ion binding / : Similarity search - Function | ||||||
| Biological species |  Desmodus rotundus (common vampire bat) Desmodus rotundus (common vampire bat) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.9 Å | ||||||
 Authors Authors | Renatus, M. / Stubbs, M.T. / Bode, W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: Catalytic domain structure of vampire bat plasminogen activator: a molecular paradigm for proteolysis without activation cleavage. Authors: Renatus, M. / Stubbs, M.T. / Huber, R. / Bringmann, P. / Donner, P. / Schleuning, W.D. / Bode, W. #1: Journal: Embo J. / Year: 1997Title: Lysine 156 Promotes the Anomalous Proenzyme Activity of Tpa: X-Ray Crystal Structure of Single-Chain Human Tpa Authors: Renatus, M. / Engh, R.A. / Stubbs, M.T. / Huber, R. / Fischer, S. / Kohnert, U. / Bode, W. #2: Journal: Curr.Opin.Struct.Biol. / Year: 1997Title: Tissue-Type Plasminogen Activator: Variants and Crystal/Solution Structures Demarcate Structural Determinants of Function Authors: Bode, W. / Renatus, M. #3: Journal: J.Mol.Biol. / Year: 1996Title: The 2.3 A Crystal Structure of the Catalytic Domain of Recombinant Two-Chain Human Tissue-Type Plasminogen Activator Authors: Lamba, D. / Bauer, M. / Huber, R. / Fischer, S. / Rudolph, R. / Kohnert, U. / Bode, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a5i.cif.gz | 67.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a5i.ent.gz | 49.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1a5i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a5/1a5iftp://data.pdbj.org/pub/pdb/validation_reports/a5/1a5i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1rtfS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29716.600 Da / Num. of mol.: 1 / Fragment: UNP residues 213-477 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Desmodus rotundus (common vampire bat) / Organ: SALIVARY GLANDS / Cell line (production host): BHK / Production host: Mesocricetus auratus (golden hamster) / References: UniProt: P98119, t-plasminogen activator |
|---|---|
| #2: Chemical | ChemComp-0GJ / 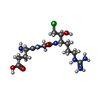  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 395.862 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28ClN6O5 / References: GLU-GLY-ARG-CHLOROMETHYL KETONE Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 395.862 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28ClN6O5 / References: GLU-GLY-ARG-CHLOROMETHYL KETONE |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Nonpolymer details | THE UNBOUND FORM OF THE INHIBITOR IS GLU-GLY-ARG-CHLOROMETHYLKETONE. UPON REACTION WITH PROTEIN IT ...THE UNBOUND FORM OF THE INHIBITOR IS GLU-GLY-ARG-CHLOROMETH |
| Sequence details | THE PROTEIN IS A DELETION MUTANT OF FULL-LENGTH DSPAALPHA1 (P98119) CONSISTING OF A SIGNAL PEPTIDE ...THE PROTEIN IS A DELETION MUTANT OF FULL-LENGTH DSPAALPHA1 (P98119) CONSISTING |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.58 Å3/Da / Density % sol: 65.66 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 9 / Details: pH 9.0 | |||||||||||||||
| Crystal | *PLUS Density % sol: 59 % | |||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, sitting drop / PH range low: 9.2 / PH range high: 8.8 | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 18, 1996 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.9→25 Å / Num. obs: 9582 / % possible obs: 97.2 % / Observed criterion σ(I): 3 / Redundancy: 2.3 % / Biso Wilson estimate: 65.48 Å2 / Rmerge(I) obs: 0.092 / Net I/σ(I): 3.51 |
| Reflection shell | Resolution: 2.9→3 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.405 / Mean I/σ(I) obs: 1.9 / % possible all: 97.9 |
| Reflection | *PLUS Num. measured all: 22337 |
| Reflection shell | *PLUS % possible obs: 97.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1RTF Resolution: 2.9→7 Å / Data cutoff high absF: 100000 / Data cutoff low absF: 0.1 / Isotropic thermal model: RESTRAINED / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.9→7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.9→3 Å / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.3489 |
