 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-52217: Cryo-EM structure of the Arabidopsis thaliana CAT4 transporter in... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the Arabidopsis thaliana CAT4 transporter in the outward-open apo state | |||||||||
 Map data Map data | Sharpened EM map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | cationic amino acid transporter / APC fold / LeuT fold / cholesterol / outward-open state / cationic amino acids / SLC7 / MEMBRANE PROTEIN | |||||||||
| Function / homology | Cationic amino acid transporter, C-terminal / C-terminus of AA_permease / plant-type vacuole membrane / Amino acid/polyamine transporter I / Amino acid permease / plant-type vacuole / amino acid transmembrane transporter activity / amino acid transport / Cationic amino acid transporter 4, vacuolar Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.27 Å | |||||||||
 Authors Authors | Kolokouris D / Kato T / Newstead S | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2019 Title: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty ...Authors: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /   Abstract: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_52217.map.gz | 59.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-52217-v30.xmlemd-52217.xml | 24.9 KB 24.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_52217_fsc.xml | 8.5 KB | Display | FSC data file |
| Images |  emd_52217.png emd_52217.png | 44.9 KB | ||
| Filedesc metadata | emd-52217.cif.gz | 7.5 KB | ||
| Others | emd_52217_additional_1.map.gzemd_52217_half_map_1.map.gzemd_52217_half_map_2.map.gz | 31.4 MB 59.4 MB 59.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-52217ftp://ftp.pdbj.org/pub/emdb/structures/EMD-52217 http://ftp.pdbj.org/pub/emdb/structures/EMD-52217ftp://ftp.pdbj.org/pub/emdb/structures/EMD-52217 | HTTPS FTP |
-Related structure data
| Related structure data |  9hjkMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_52217.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Sharpened EM map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.832 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Unsharpened EM map
| File | emd_52217_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened EM map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #2
| File | emd_52217_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_52217_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Complex of Arabidopsis thaliana cationic amino acid transporter 4...
| Entire | Name: Complex of Arabidopsis thaliana cationic amino acid transporter 4 with a synthetic nanobody and cholesterol |
|---|---|
| Components |
|
-Supramolecule #1: Complex of Arabidopsis thaliana cationic amino acid transporter 4...
| Supramolecule | Name: Complex of Arabidopsis thaliana cationic amino acid transporter 4 with a synthetic nanobody and cholesterol type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: #2, #1 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Cationic amino acid transporter 4, vacuolar
| Macromolecule | Name: Cationic amino acid transporter 4, vacuolar / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 59.39016 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: PHQLAKKLSA VDLVAIGVGT TIGAGVYILV GTVAREHTGP ALAVSFFIAG VAAALSACCY AELASRCPSA GSAYHYAYIC LGEGIAWLV GWALVLDYTI GGSAIARGIT PNLASFFGGL DNLPVFLARQ TIPGVGIVVD PCAALLIMIV TILLCFGIKE S STVQAIVT ...String: PHQLAKKLSA VDLVAIGVGT TIGAGVYILV GTVAREHTGP ALAVSFFIAG VAAALSACCY AELASRCPSA GSAYHYAYIC LGEGIAWLV GWALVLDYTI GGSAIARGIT PNLASFFGGL DNLPVFLARQ TIPGVGIVVD PCAALLIMIV TILLCFGIKE S STVQAIVT SVNVCTLVFI IVVGGYLACK TGWVGYDLPS GYFPFGLNGI LAGSAVVFFS YIGFDTVTST AEEVKNPQRD LP LGIGIAL LICCILYMLL SVVIVGLVPY YSLNPDTPIS SAFGDSGMQW AAYILTTGAI TALCASLLGS LLAQPRIFMA MAR DGLLPA FFSEISPRTQ VPVKSTIAIG VLAAALAFFM DVAQLSEMVS VGTLMAFTAV AVCVLVLRYV PPDGVPLSSS SQTL SDTDE SRAETENFLV DAIESSDSPL LGNETARDEK YFGKRRKIAA WSIALVCIGV LGLASAASAE RLPSFPRFTI CGVSA VILL GSLITLGYID EDEERHNFGH KGGFLCPFVP YLPVLCILIN TYLIINIGAG TWIRVLIWLL IGSMIYIFYG RSHSLL NN UniProtKB: Cationic amino acid transporter 4, vacuolar |
-Macromolecule #2: SybB5
| Macromolecule | Name: SybB5 / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 12.826291 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: GSSSQVQLVE SGGGLVQAGG SLRLSCAASG FPVNMYWMHW YRQAPGKERE WVAAIQSYGQ WTAYADSVKG RFTISRDNAK NTVYLQMNS LKPEDTAVYY CAVGVGGYYL GQGTQVTVS |
-Macromolecule #3: CHOLESTEROL
| Macromolecule | Name: CHOLESTEROL / type: ligand / ID: 3 / Number of copies: 1 / Formula: CLR |
|---|---|
| Molecular weight | Theoretical: 386.654 Da |
| Chemical component information | 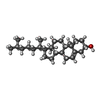 ChemComp-CLR: |
-Macromolecule #4: Lauryl Maltose Neopentyl Glycol
| Macromolecule | Name: Lauryl Maltose Neopentyl Glycol / type: ligand / ID: 4 / Number of copies: 1 / Formula: AV0 |
|---|---|
| Molecular weight | Theoretical: 1.005188 KDa |
| Chemical component information |  ChemComp-AV0: |
-Macromolecule #5: water
| Macromolecule | Name: water / type: ligand / ID: 5 / Number of copies: 1 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3.4 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 20 mM Tris-HCl, 150 mM NaCl, 0.003% w/v LMNG |
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV |
| Details | The sample was homogeneously monodisperse. |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Average exposure time: 3.0 sec. / Average electron dose: 58.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: AlphaFold / Chain - Initial model type: in silico model |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: cross-correlation coefficient |
| Output model | PDB-9hjk: |

