Movie
Movie Controller
Controller Sample components
Sample components

[English] 日本語
 Yorodumi
Yorodumi- EMDB-5172: 3.6 Angstrom cryoEM structure of human adenovirus type 5 (obsolet... -
+ Open data
Open data

- Basic information
Basic information

 Sample
Sample Citation
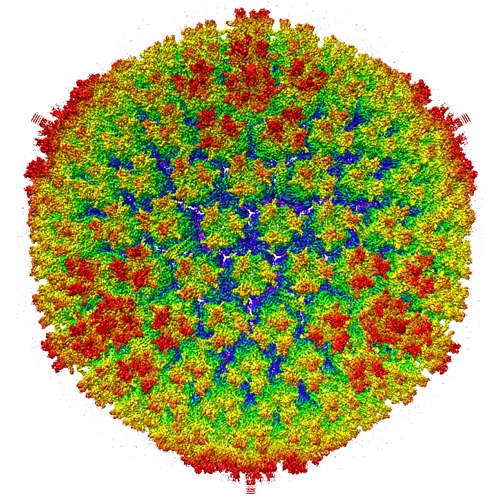
Citation- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Header (meta data) |  EMDB header EMDB header | |||
|---|---|---|---|---|
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5172ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5172 http://ftp.pdbj.org/pub/emdb/structures/EMD-5172ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5172 | HTTPS FTP |
-Validation report
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-5172ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-5172 | HTTPS FTP |
|---|
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
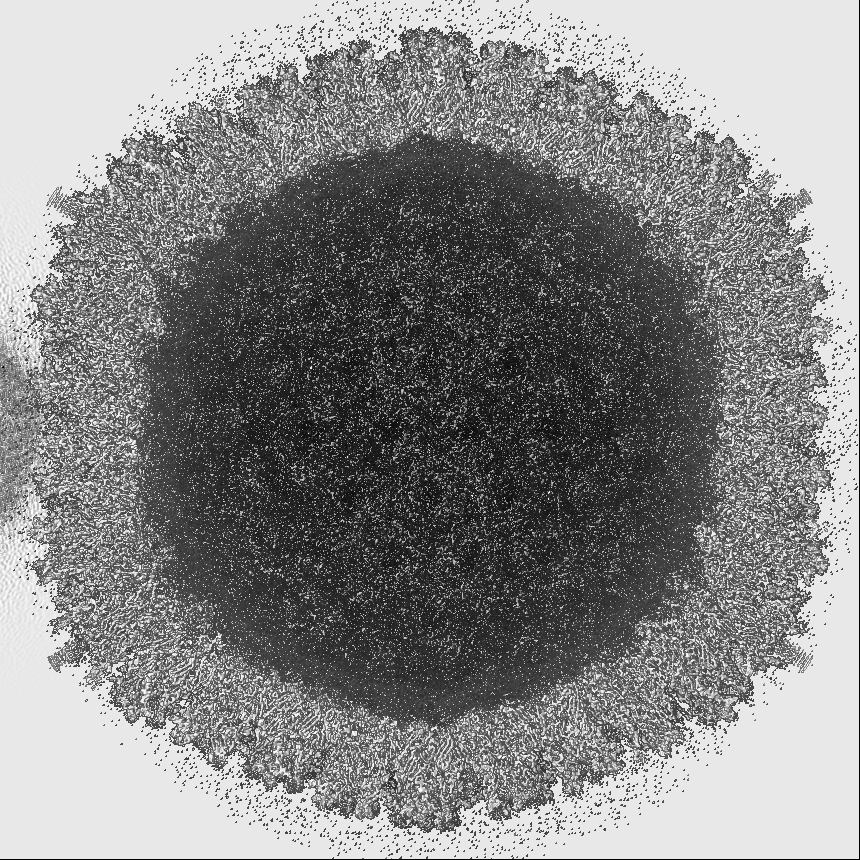
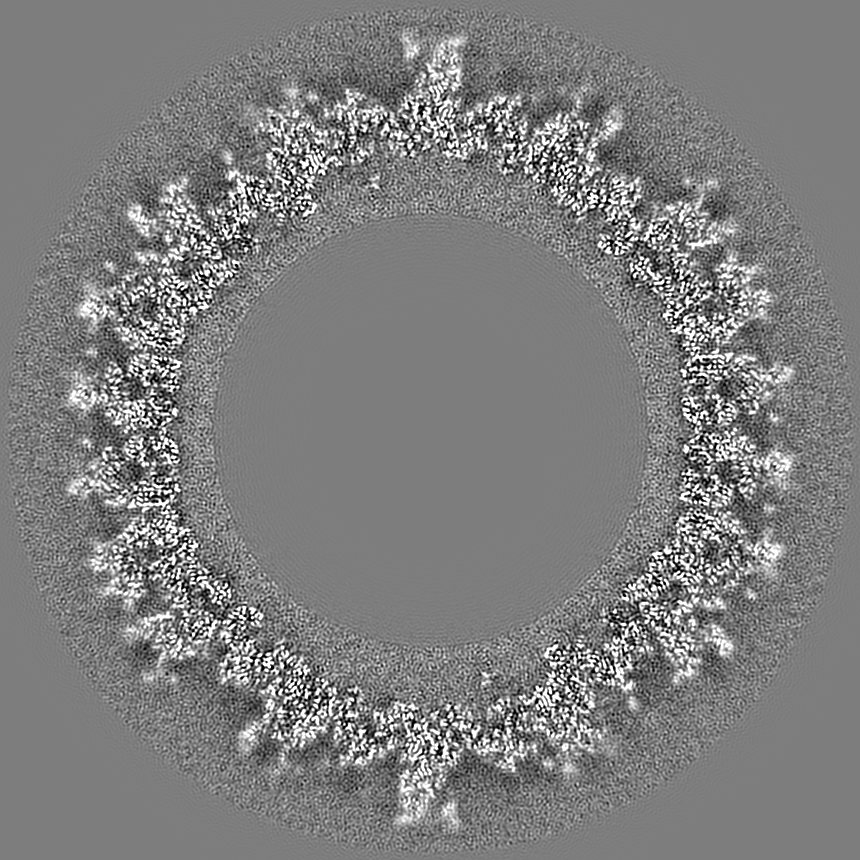
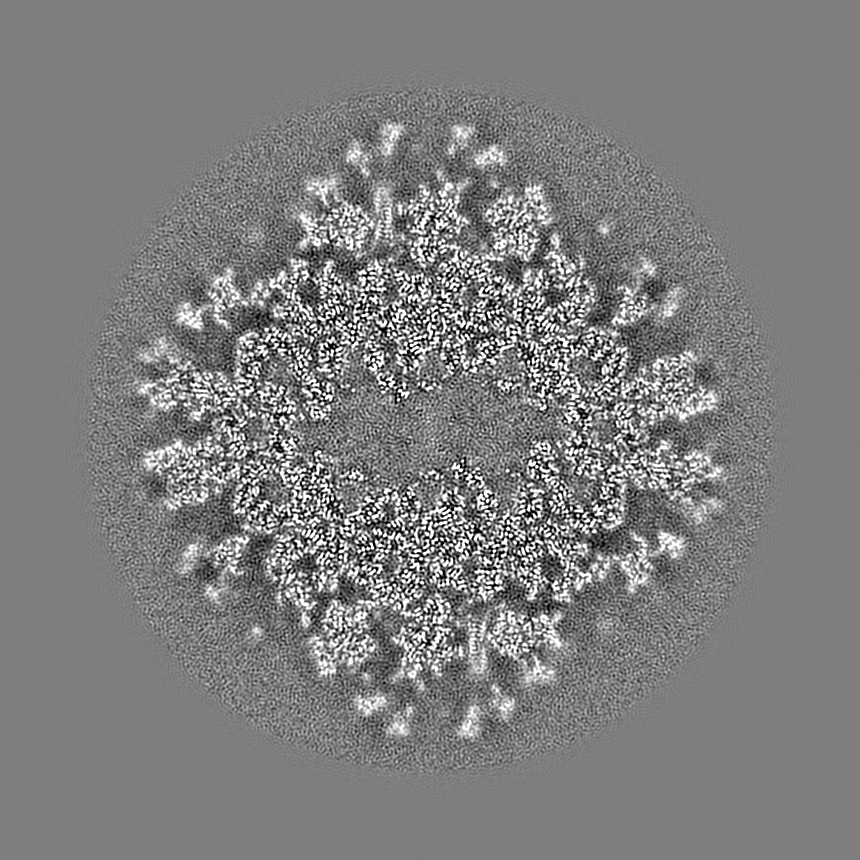
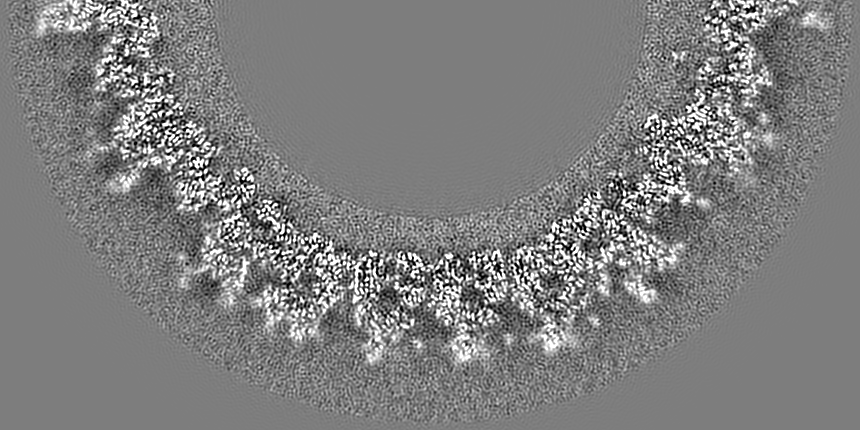
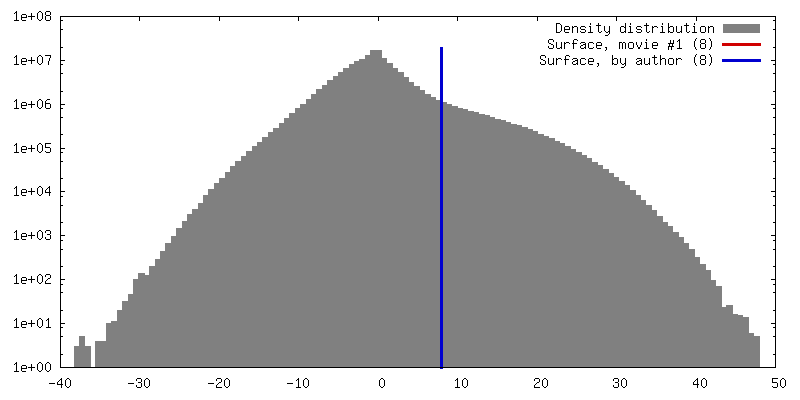
- Map
Map
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Voxel size | X=Y=Z: 0 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 (Sec.)
(Sec.) (Row.)
(Row.) (Col.)
(Col.)