 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the Retron IA Complex without the HNH Nuclease | |||||||||
 Map data Map data | Structure of the Retron IA Complex without the HNH Nuclease | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Retron / IA / Immune / Transferase-DNA-RNA complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationDNA synthesis involved in DNA repair / RNA-directed DNA polymerase / RNA-directed DNA polymerase activity / double-strand break repair / defense response to virus / RNA binding / ATP binding / metal ion binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.99 Å | |||||||||
 Authors Authors | Burman N / Thomas-George J / Wilkinson R / Wiedenheft B | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: bioRxiv / Year: 2025 Title: Structural basis of antiphage defense by an ATPase-associated reverse transcriptase. Authors: Jerrin Thomas George / Nathaniel Burman / Royce A Wilkinson / Senuri de Silva / Quynh McKelvey-Pham / Murat Buyukyoruk / Adelaide Dale / Hannah Landman / Ava Graham / Steven Z DeLuca / Blake Wiedenheft / Abstract: Reverse transcriptases (RTs) have well-established roles in the replication and spread of retroviruses and retrotransposons. However, recent evidence suggests that RTs have been conscripted by cells ...Reverse transcriptases (RTs) have well-established roles in the replication and spread of retroviruses and retrotransposons. However, recent evidence suggests that RTs have been conscripted by cells for diverse roles in antiviral defense. Here we determine structures of a type I-A retron, which explain how RNA, DNA, RT, HNH-nuclease and four molecules of an SMC-family ATPase assemble into a 364 kDa complex that provides phage defense. We show that phage-encoded nucleases trigger degradation of the retron-associated DNA, leading to disassembly of the retron and activation of the HNH nuclease. The HNH nuclease cleaves tRNA, stalling protein synthesis and arresting viral replication. Taken together, these data reveal diverse and paradoxical roles for RTs in the perpetuation and elimination of genetic parasites. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_49056.map.gz | 211.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-49056-v30.xmlemd-49056.xml | 29.4 KB 29.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_49056_fsc.xml | 15.9 KB | Display | FSC data file |
| Images |  emd_49056.png emd_49056.png | 47.5 KB | ||
| Filedesc metadata | emd-49056.cif.gz | 8.8 KB | ||
| Others | emd_49056_half_map_1.map.gzemd_49056_half_map_2.map.gz | 391.8 MB 391.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-49056ftp://ftp.pdbj.org/pub/emdb/structures/EMD-49056 http://ftp.pdbj.org/pub/emdb/structures/EMD-49056ftp://ftp.pdbj.org/pub/emdb/structures/EMD-49056 | HTTPS FTP |
-Related structure data
| Related structure data |  9n6cMC  9n69C  9n6bC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_49056.map.gz / Format: CCP4 / Size: 421.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of the Retron IA Complex without the HNH Nuclease | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.9061 Å | ||||||||||||||||||||||||||||||||||||
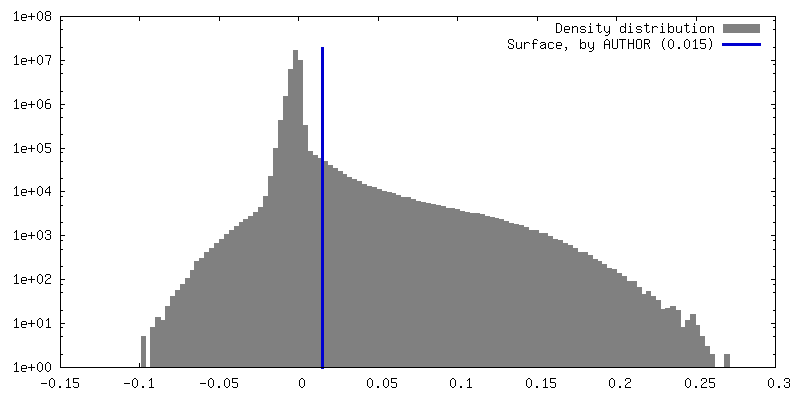
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: Half Map B
| File | emd_49056_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half Map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
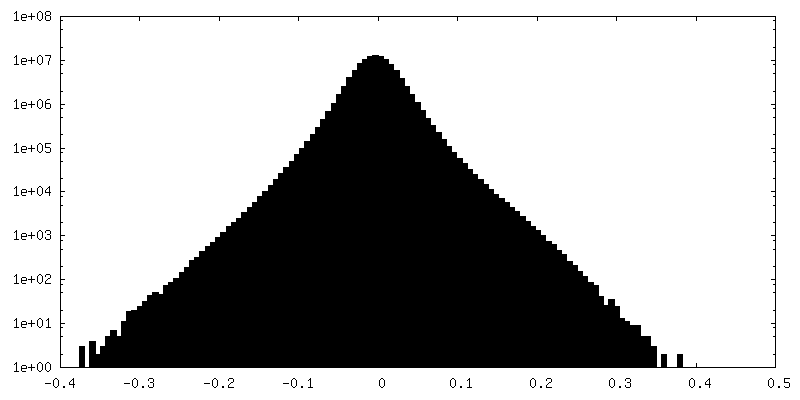
| Density Histograms |






-Half map: Half Map A
| File | emd_49056_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half Map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
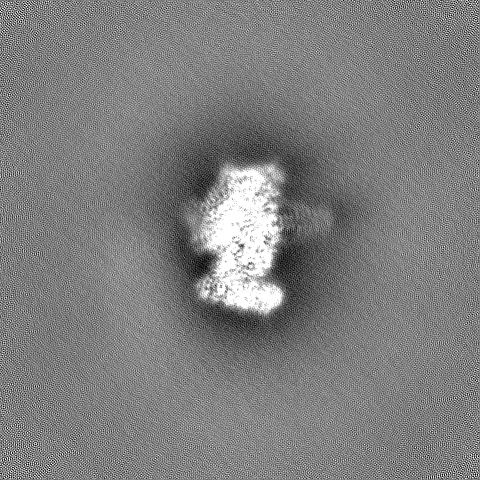



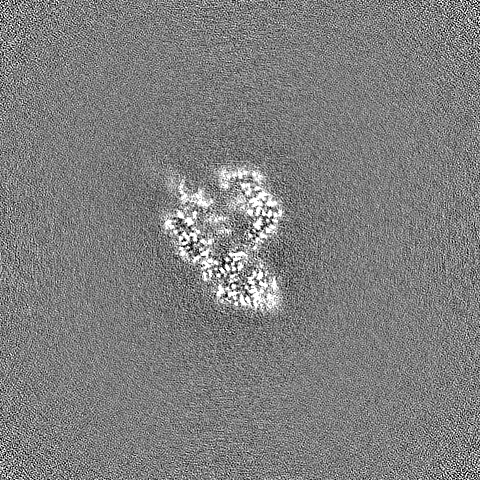


- Sample components
Sample components
-Entire : Retron IA surveillance complex with HNH nuclease bound in the "do...
| Entire | Name: Retron IA surveillance complex with HNH nuclease bound in the "down" orientation |
|---|---|
| Components |
|
-Supramolecule #1: Retron IA surveillance complex with HNH nuclease bound in the "do...
| Supramolecule | Name: Retron IA surveillance complex with HNH nuclease bound in the "down" orientation type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#4 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: AAA family ATPase
| Macromolecule | Name: AAA family ATPase / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 63.393953 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MWSHPQFEKI NKMNLETCYV DFLELESHVI NEDYLKESVE LQKLISTLNE SKFHLNKIGI HDFKRIRELQ ISLEDDLTVF VGDNGFGKS TILDAIAIVL SWLRSNIEKE SKPGTYIKSH EVNNSVDVEY ASIDANIKLK DFNTSILITK AKEGAYYSRN N ELLGVKKL ...String: MWSHPQFEKI NKMNLETCYV DFLELESHVI NEDYLKESVE LQKLISTLNE SKFHLNKIGI HDFKRIRELQ ISLEDDLTVF VGDNGFGKS TILDAIAIVL SWLRSNIEKE SKPGTYIKSH EVNNSVDVEY ASIDANIKLK DFNTSILITK AKEGAYYSRN N ELLGVKKL ASIYRLVNKY VDNASLPLMA YYSIARSYIG GGVDRKRKNA KTKTVWSKFD VYDEIEFDRN DFTDFFQWLV FL HNRASQE KLSESQTTIN ALFSDIQSLK ATLTQLSAID NIDSTVIKGL ELSLKEKLNY MKSLQSGEHK FNNAVSLYDS VIN TILKFL PEFQWIKLVY GDDDYKIILK KGEVELDIQQ LSQGEKTIFT LVGDLARRLI LLNPNLSNPL LGYGIVLIDE IDLH LHPQW QQTIIERLTS TFPNVQFVIT THSPQVLSTV SSRSVRILQE VEVDGVNDLI VSHPDYQIKG VSNQDALLYG MRTDP IPST KENGWLEEYK KLVELNRYSS DEALLLREKV IKHFGLDHPL VQECDDLISV LEFKNKINQH FSGSKDVK UniProtKB: AAA family ATPase |
-Macromolecule #2: RNA-directed DNA polymerase
| Macromolecule | Name: RNA-directed DNA polymerase / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO / EC number: RNA-directed DNA polymerase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 36.046191 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MQLTSKIISK FNYNRLAFQL LLNEAPKKYK VYYIPKRGAG FRVIAQPTKE LKNVQRFIVS LLQPKLPVHH KAMAYEYKKS IKDNALLHK DNNYILKMDF QNFFNKIKPD IFFSKLENTG LKLDSFDENT LRNLLFWRPG KKRSTTLILS VGAPSSPFIS N FVMYDFDK ...String: MQLTSKIISK FNYNRLAFQL LLNEAPKKYK VYYIPKRGAG FRVIAQPTKE LKNVQRFIVS LLQPKLPVHH KAMAYEYKKS IKDNALLHK DNNYILKMDF QNFFNKIKPD IFFSKLENTG LKLDSFDENT LRNLLFWRPG KKRSTTLILS VGAPSSPFIS N FVMYDFDK SLDDWCRNNG ITYSRYADDI TFSTNIKDIL CRVPKVVKKM LSLHVPGLSI NESKTIFTSM AHNRHVTGVT LT PQGNLSI GRDRKRMLFA KIHKYSLGLL SSEEINKTKG MIAFANYLEG DFLLRLQKKY GCELITKFLM EGNK UniProtKB: RNA-directed DNA polymerase |
-Macromolecule #3: Retron IA msDNA
| Macromolecule | Name: Retron IA msDNA / type: dna / ID: 3 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 28.334156 KDa |
| Sequence | String: (DT)(DA)(DA)(DA)(DG)(DA)(DC)(DA)(DG)(DC) (DG)(DA)(DA)(DA)(DG)(DA)(DC)(DA)(DC)(DA) (DG)(DA)(DT)(DT)(DT)(DC)(DT)(DC)(DC) (DT)(DT)(DC)(DG)(DC)(DA)(DT)(DA)(DT)(DC) (DT) (DG)(DC)(DC)(DC)(DC)(DG) ...String: (DT)(DA)(DA)(DA)(DG)(DA)(DC)(DA)(DG)(DC) (DG)(DA)(DA)(DA)(DG)(DA)(DC)(DA)(DC)(DA) (DG)(DA)(DT)(DT)(DT)(DC)(DT)(DC)(DC) (DT)(DT)(DC)(DG)(DC)(DA)(DT)(DA)(DT)(DC) (DT) (DG)(DC)(DC)(DC)(DC)(DG)(DG)(DG) (DC)(DA)(DG)(DG)(DG)(DA)(DT)(DG)(DC)(DG) (DA)(DA) (DG)(DG)(DA)(DG)(DA)(DA)(DA) (DT)(DC)(DT)(DG)(DT)(DG)(DT)(DC)(DT)(DT) (DT)(DC)(DG) (DC)(DA)(DA)(DC)(DC)(DC) (DT)(DA)(DA)(DA)(DC)(DC) GENBANK: GENBANK: CP026641.1 |
-Macromolecule #4: Retron IA ncRNA
| Macromolecule | Name: Retron IA ncRNA / type: rna / ID: 4 / Number of copies: 1 |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 20.258928 KDa |
| Sequence | String: UAGUGUAGGA ACAUUGGUUC CAGCCGGGUG AUUAGCCAGG CUUAAAUUUA UUGUCCGGUU UAG GENBANK: GENBANK: CP026641.1 |
-Macromolecule #5: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 5 / Number of copies: 3 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Grid | Model: C-flat-1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 45 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 1 / Number real images: 13695 / Average electron dose: 56.69 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 45000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | Chain - Source name: AlphaFold / Chain - Initial model type: in silico model Details: The structure of each subunit was predicted individually in Alphafold before rigid body fitting in ChimeraX |
|---|---|
| Details | Alphafold predicted structures of each individual subunit were first rigid body fit in ChimeraX using the fit in map command before initial relaxation using ISOLDE and final refinement in PHENIX RealSpaceRefinement. COOT was used for manual editing. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Cross-correlation coefficient |
| Output model | PDB-9n6c: |
