 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-44534: Human DNA polymerase theta helicase domain tetramer in the apo form -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human DNA polymerase theta helicase domain tetramer in the apo form | |||||||||
 Map data Map data | Combined tetramer map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | DNA repair / TMEJ / MMEJ / DNA binding protein | |||||||||
| Function / homology |  Function and homology information Function and homology informationdouble-strand break repair via alternative nonhomologous end joining / HDR through MMEJ (alt-NHEJ) / single-stranded DNA helicase activity / DNA synthesis involved in DNA repair / replication fork processing / mitochondrial nucleoid / 5'-deoxyribose-5-phosphate lyase activity / error-prone translesion synthesis / negative regulation of double-strand break repair via homologous recombination / somatic hypermutation of immunoglobulin genes ...double-strand break repair via alternative nonhomologous end joining / HDR through MMEJ (alt-NHEJ) / single-stranded DNA helicase activity / DNA synthesis involved in DNA repair / replication fork processing / mitochondrial nucleoid / 5'-deoxyribose-5-phosphate lyase activity / error-prone translesion synthesis / negative regulation of double-strand break repair via homologous recombination / somatic hypermutation of immunoglobulin genes / site of DNA damage / DNA helicase activity / protein homooligomerization / base-excision repair / RNA-directed DNA polymerase / RNA-directed DNA polymerase activity / double-strand break repair / site of double-strand break / DNA-directed DNA polymerase / DNA helicase / damaged DNA binding / DNA-directed DNA polymerase activity / DNA repair / chromatin binding / DNA damage response / magnesium ion binding / Golgi apparatus / ATP hydrolysis activity / nucleoplasm / ATP binding / identical protein binding / nucleus / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.3 Å | |||||||||
 Authors Authors | Zerio CJ / Lander GC | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2025 Title: Human polymerase θ helicase positions DNA microhomologies for double-strand break repair. Authors: Christopher J Zerio / Yonghong Bai / Brian A Sosa-Alvarado / Timothy Guzi / Gabriel C Lander / Abstract: DNA double-strand breaks occur daily in all human cells and must be repaired with high fidelity to minimize genomic instability. Deficiencies in high-fidelity DNA repair by homologous recombination ...DNA double-strand breaks occur daily in all human cells and must be repaired with high fidelity to minimize genomic instability. Deficiencies in high-fidelity DNA repair by homologous recombination lead to dependence on DNA polymerase θ, which identifies DNA microhomologies in 3' single-stranded DNA overhangs and anneals them to initiate error-prone double-strand break repair. The resulting genomic instability is associated with numerous cancers, thereby making this polymerase an attractive therapeutic target. However, despite the biomedical importance of polymerase θ, the molecular details of how it initiates DNA break repair remain unclear. Here, we present cryo-electron microscopy structures of the polymerase θ helicase domain bound to microhomology-containing DNA, revealing DNA-induced rearrangements of the helicase that enable DNA repair. Our structures show that DNA-bound helicase dimers facilitate a microhomology search that positions 3' single-stranded DNA ends in proximity to align complementary bases and anneal DNA microhomology. We characterize the molecular determinants that enable the helicase domain of polymerase θ to identify and pair DNA microhomologies to initiate mutagenic DNA repair, thereby providing insight into potentially targetable interactions for therapeutic interventions. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_44534.map.gz | 7.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-44534-v30.xmlemd-44534.xml | 27.7 KB 27.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_44534_fsc.xml | 12 KB | Display | FSC data file |
| Images | 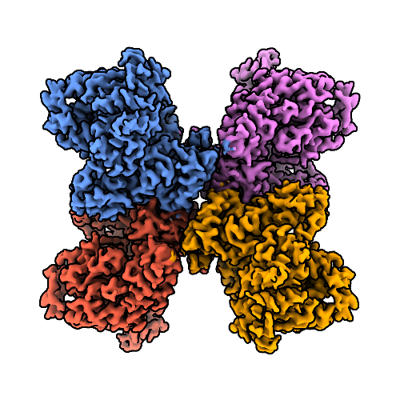 emd_44534.png emd_44534.png | 163.4 KB | ||
| Masks | emd_44534_msk_1.map | 125 MB | Mask map | |
| Filedesc metadata | emd-44534.cif.gz | 7.7 KB | ||
| Others | emd_44534_additional_1.map.gzemd_44534_additional_2.map.gzemd_44534_half_map_1.map.gzemd_44534_half_map_2.map.gz | 118 MB 117.8 MB 115.8 MB 115.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-44534ftp://ftp.pdbj.org/pub/emdb/structures/EMD-44534 http://ftp.pdbj.org/pub/emdb/structures/EMD-44534ftp://ftp.pdbj.org/pub/emdb/structures/EMD-44534 | HTTPS FTP |
-Related structure data
| Related structure data |  9bh6MC  9bh7C  9bh8C  9bh9C  9bhaC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_44534.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Combined tetramer map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.833 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_44534_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: Locally refined protomer map
| File | emd_44534_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Locally refined protomer map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Additional map: D2 symmetric tetramer map
| File | emd_44534_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | D2 symmetric tetramer map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Protomer half map A
| File | emd_44534_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Protomer half map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





-Half map: Protomer half map B
| File | emd_44534_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Protomer half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

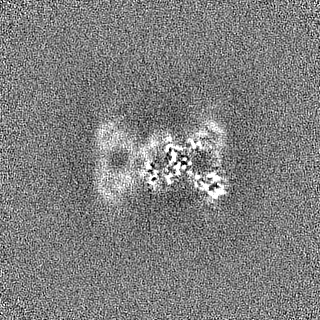



- Sample components
Sample components
-Entire : Human DNA polymerase theta helicase domain
| Entire | Name: Human DNA polymerase theta helicase domain |
|---|---|
| Components |
|
-Supramolecule #1: Human DNA polymerase theta helicase domain
| Supramolecule | Name: Human DNA polymerase theta helicase domain / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 400 KDa |
-Macromolecule #1: DNA polymerase theta
| Macromolecule | Name: DNA polymerase theta / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO / EC number: DNA helicase |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 99.671344 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: NLLRRSGKRR RSESGSDSFS GSGGDSSASP QFLSGSVLSP PPGLGRCLKA AAAGECKPTV PDYERDKLLL ANWGLPKAVL EKYHSFGVK KMFEWQAECL LLGQVLEGKN LVYSAPTSAG KTLVAELLIL KRVLEMRKKA LFILPFVSVA KEKKYYLQSL F QEVGIKVD ...String: NLLRRSGKRR RSESGSDSFS GSGGDSSASP QFLSGSVLSP PPGLGRCLKA AAAGECKPTV PDYERDKLLL ANWGLPKAVL EKYHSFGVK KMFEWQAECL LLGQVLEGKN LVYSAPTSAG KTLVAELLIL KRVLEMRKKA LFILPFVSVA KEKKYYLQSL F QEVGIKVD GYMGSTSPSR HFSSLDIAVC TIERANGLIN RLIEENKMDL LGMVVVDELH MLGDSHRGYL LELLLTKICY IT RKSASCQ ADLASSLSNA VQIVGMSATL PNLELVASWL NAELYHTDFR PVPLLESVKV GNSIYDSSMK LVREFEPMLQ VKG DEDHVV SLCYETICDN HSVLLFCPSK KWCEKLADII AREFYNLHHQ AEGLVKPSEC PPVILEQKEL LEVMDQLRRL PSGL DSVLQ KTVPWGVAFH HAGLTFEERD IIEGAFRQGL IRVLAATSTL SSGVNLPARR VIIRTPIFGG RPLDILTYKQ MVGRA GRKG VDTVGESILI CKNSEKSKGI ALLQGSLKPV RSCLQRREGE EVTGSMIRAI LEIIVGGVAS TSQDMHTYAA CTFLAA SMK EGKQGIQRNQ ESVQLGAIEA CVMWLLENEF IQSTEASDGT EGKVYHPTHL GSATLSSSLS PADTLDIFAD LQRAMKG FV LENDLHILYL VTPMFEDWTT IDWYRFFCLW EKLPTSMKRV AELVGVEEGF LARCVKGKVV ARTERQHRQM AIHKRFFT S LVLLDLISEV PLREINQKYG CNRGQIQSLQ QSAAVYAGMI TVFSNRLGWH NMELLLSQFQ KRLTFGIQRE LCDLVRVSL LNAQRARVLY ASGFHTVADL ARANIVEVEV ILKNAVPFKS ARKAVDEEEE AVEERRNMRT IWVTGRKGLT EREAAALIVE EARMILQQD LVEM UniProtKB: DNA polymerase theta |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.9 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. Details: Grids were glow discharged under vacuum for 30 s at 15 mA in a Pelco easiGlow 91000 Glow Discharge Cleaning System. | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: HOMEMADE PLUNGER Details: 3 microliters of sample was applied to the surface of the grid, blotted with Whatman 1 filter paper until 2 seconds after the liquid spot on the filter paper stopped spreading, and the grid ...Details: 3 microliters of sample was applied to the surface of the grid, blotted with Whatman 1 filter paper until 2 seconds after the liquid spot on the filter paper stopped spreading, and the grid was plunged into a liquid ethane pool cooled by liquid nitrogen using a manual plunge freezer.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Temperature | Min: 70.0 K / Max: 77.0 K |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 3906 / Average exposure time: 0.9 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 150.0 µm / Calibrated defocus max: 2.0 µm / Calibrated defocus min: 0.3 µm / Calibrated magnification: 60024 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

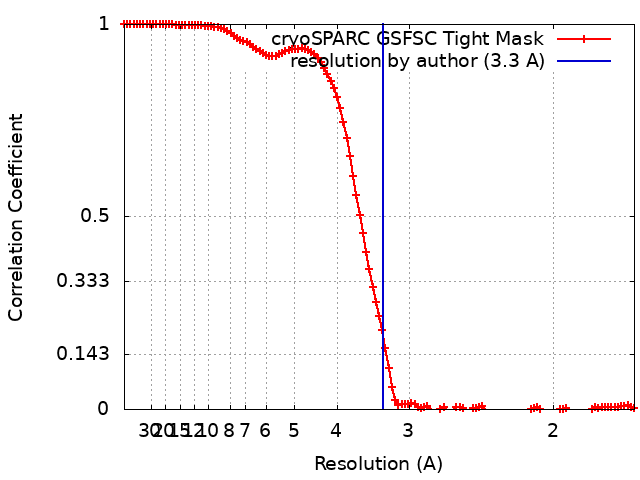
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 67-892 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | We used ISOLDE to flexibly fit one protomer into the locally refined protomer map. The output protomer model was multiplied and we used ISOLDE to fit four protomers into the tetramer map. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 54.43 / Target criteria: Cross-correlation coefficient |
| Output model | PDB-9bh6: |
