 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-42974: Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral sy... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral symmetry | |||||||||||||||
 Map data Map data | ||||||||||||||||
 Sample Sample |
| |||||||||||||||
 Keywords Keywords | Encapsulin / VIRUS LIKE PARTICLE | |||||||||||||||
| Function / homology | Type 1 encapsulin shell protein / Encapsulating protein for peroxidase / : / encapsulin nanocompartment / iron ion transport / intracellular iron ion homeostasis / Type 1 encapsulin shell protein EncA Function and homology information Function and homology information | |||||||||||||||
| Biological species |  Myxococcus xanthus DK 1622 (bacteria) / Myxococcus xanthus (bacteria) Myxococcus xanthus DK 1622 (bacteria) / Myxococcus xanthus (bacteria) | |||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.43 Å | |||||||||||||||
 Authors Authors | Szyszka TN / Andreas MP / Lie F / Miller LM / Adamson LSR / Fatehi F / Twarock R / Draper BE / Jarrold MF / Giessen TW / Lau YH | |||||||||||||||
| Funding support |  Australia, Australia,  United States, 4 items United States, 4 items
| |||||||||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2024 Title: Point mutation in a virus-like capsid drives symmetry reduction to form tetrahedral cages. Authors: Taylor N Szyszka / Michael P Andreas / Felicia Lie / Lohra M Miller / Lachlan S R Adamson / Farzad Fatehi / Reidun Twarock / Benjamin E Draper / Martin F Jarrold / Tobias W Giessen / Yu Heng Lau /  Abstract: Protein capsids are a widespread form of compartmentalization in nature. Icosahedral symmetry is ubiquitous in capsids derived from spherical viruses, as this geometry maximizes the internal volume ...Protein capsids are a widespread form of compartmentalization in nature. Icosahedral symmetry is ubiquitous in capsids derived from spherical viruses, as this geometry maximizes the internal volume that can be enclosed within. Despite the strong preference for icosahedral symmetry, we show that simple point mutations in a virus-like capsid can drive the assembly of unique symmetry-reduced structures. Starting with the encapsulin from , a 180-mer bacterial capsid that adopts the well-studied viral HK97 fold, we use mass photometry and native charge detection mass spectrometry to identify a triple histidine point mutant that forms smaller dimorphic assemblies. Using cryoelectron microscopy, we determine the structures of a precedented 60-mer icosahedral assembly and an unexpected 36-mer tetrahedron that features significant geometric rearrangements around a new interaction surface between capsid protomers. We subsequently find that the tetrahedral assembly can be generated by triple-point mutation to various amino acids and that even a single histidine point mutation is sufficient to form tetrahedra. These findings represent a unique example of tetrahedral geometry when surveying all characterized encapsulins, HK97-like capsids, or indeed any virus-derived capsids reported in the Protein Data Bank, revealing the surprising plasticity of capsid self-assembly that can be accessed through minimal changes in the protein sequence. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
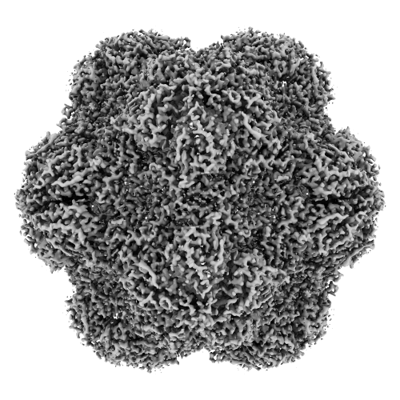
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_42974.map.gz | 200.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-42974-v30.xmlemd-42974.xml | 19.5 KB 19.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_42974_fsc.xml | 12.6 KB | Display | FSC data file |
| Images | 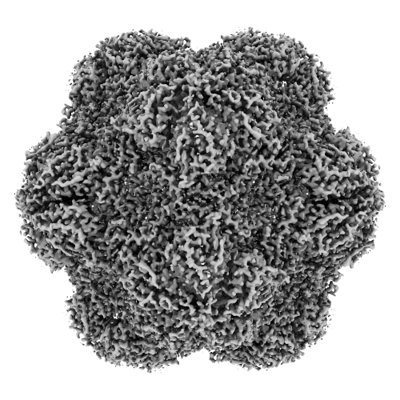 emd_42974.png emd_42974.png | 158.4 KB | ||
| Filedesc metadata | emd-42974.cif.gz | 6.5 KB | ||
| Others | emd_42974_half_map_1.map.gzemd_42974_half_map_2.map.gz | 199.9 MB 199.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-42974ftp://ftp.pdbj.org/pub/emdb/structures/EMD-42974 http://ftp.pdbj.org/pub/emdb/structures/EMD-42974ftp://ftp.pdbj.org/pub/emdb/structures/EMD-42974 | HTTPS FTP |
-Related structure data
| Related structure data |  8v4nMC  8v4qC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_42974.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
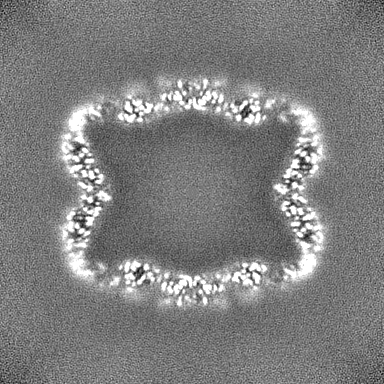
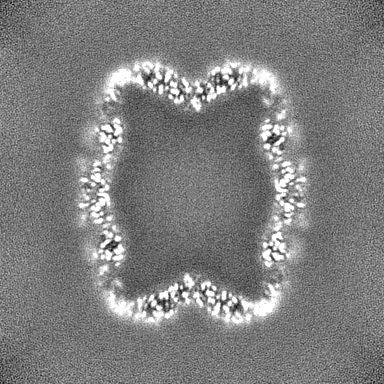
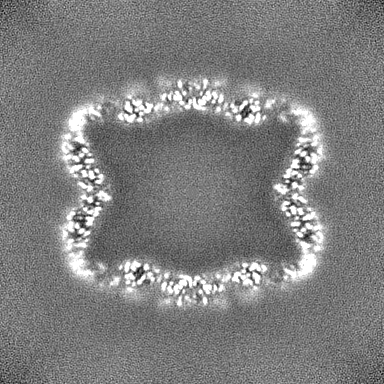
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.867 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) X (Row.)
X (Row.) Y (Col.)
Y (Col.)





















-Supplemental data
-Half map: #1
| File | emd_42974_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
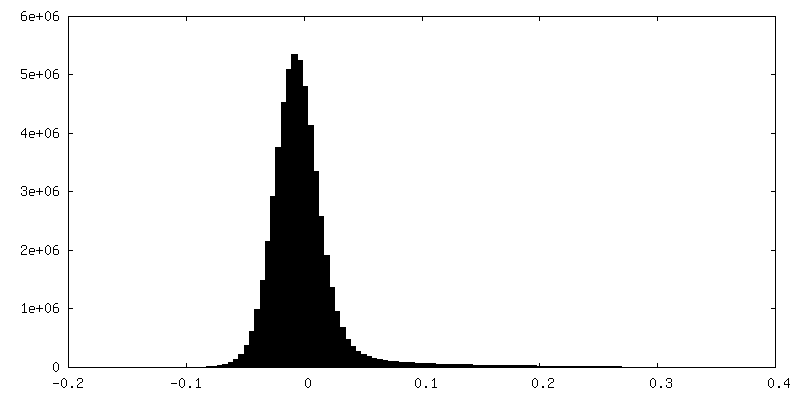
| Density Histograms |






-Half map: #2
| File | emd_42974_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral sy...
| Entire | Name: Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral symmetry |
|---|---|
| Components |
|
-Supramolecule #1: Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral sy...
| Supramolecule | Name: Myxococcus xanthus EncA 3xHis pore mutant with T=1 icosahedral symmetry type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Myxococcus xanthus DK 1622 (bacteria) |
| Molecular weight | Theoretical: 1.9 MDa |
-Macromolecule #1: Type 1 encapsulin shell protein EncA
| Macromolecule | Name: Type 1 encapsulin shell protein EncA / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Myxococcus xanthus (bacteria) / Strain: DK1622 |
| Molecular weight | Theoretical: 31.819082 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MPDFLGHAEN PLREEEWARL NETVIQVARR SLVGRRILDI YGPLGAGVQT VPYDEFQGVS PGAVDIVGEQ ETAMVFTDAR KFKTIPIIY KDFLLHWRDI EAARTHNMPL DVSAAAGAAA LCAQQEDELI FYGDARLGYE GLMTANGRLT VPLGDWTSPG G GFQAIVEA ...String: MPDFLGHAEN PLREEEWARL NETVIQVARR SLVGRRILDI YGPLGAGVQT VPYDEFQGVS PGAVDIVGEQ ETAMVFTDAR KFKTIPIIY KDFLLHWRDI EAARTHNMPL DVSAAAGAAA LCAQQEDELI FYGDARLGYE GLMTANGRLT VPLGDWTSPG G GFQAIVEA TRKLNEQGHF GPYAVVLSPR LYSQLHRIYE HHHVLEIETI RQLASDGVYQ SNRLRGESGV VVSTGRENMD LA VSMDMVA AYLGASRMNH PFRVLEALLL RIKHPDAICT LEGAGATERR UniProtKB: Type 1 encapsulin shell protein EncA |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3.0 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
Details: 50 mM Tris pH 8.0, 200 mM NaCl | |||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Details: 5 mA for 60 seconds under vacuum | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 295 K / Instrument: FEI VITROBOT MARK IV / Details: Blot force: 5 Blot time: 2 seconds. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 4092 pixel / Digitization - Dimensions - Height: 5760 pixel / Number grids imaged: 1 / Number real images: 1475 / Average exposure time: 2.0972 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | Chain - Source name: Other / Chain - Initial model type: in silico model / Details: ESMfold was used to make starting model |
|---|---|
| Details | The starting model was generated using ESMfold then fit into the map using ChimeraX. The placed coordinates were then manually refined using Coot, followed by real space refinement in Phenix. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Overall B value: 98.5 / Target criteria: Cross-correlation coefficient |
| Output model | PDB-8v4n: |
