 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human RADX tetramer bound to ssDNA | |||||||||
 Map data Map data | RADX tetramer bound to ssDNA dT25 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | oligomer OB-fold / RAD51 regulator / DNA BINDING PROTEIN / DNA BINDING PROTEIN-DNA complex | |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of double-strand break repair via homologous recombination / regulation of DNA repair / replication fork / single-stranded DNA binding / nuclear speck / RNA binding Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
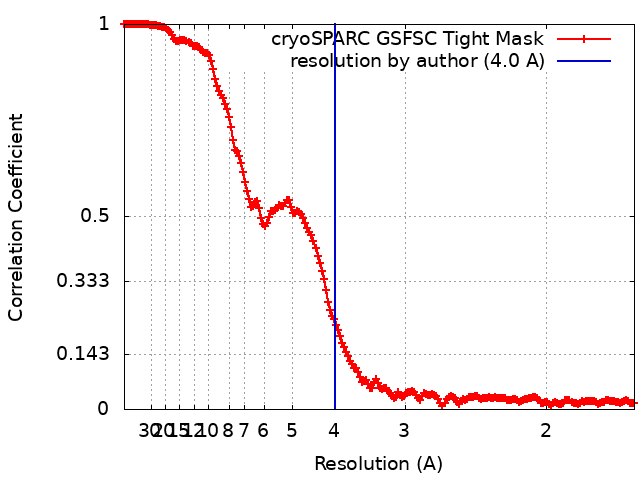
| Method | single particle reconstruction / cryo EM / Resolution: 4.0 Å | |||||||||
 Authors Authors | Balakrishnan S / Chazin WJ | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2024 Title: Structure of RADX and mechanism for regulation of RAD51 nucleofilaments. Authors: Swati Balakrishnan / Madison Adolph / Miaw-Sheue Tsai / Tae Akizuki / Kaitlyn Gallagher / David Cortez / Walter J Chazin / Abstract: Replication fork reversal is a fundamental process required for resolution of encounters with DNA damage. A key step in the stabilization and eventual resolution of reversed forks is formation of ...Replication fork reversal is a fundamental process required for resolution of encounters with DNA damage. A key step in the stabilization and eventual resolution of reversed forks is formation of RAD51 nucleoprotein filaments on exposed single strand DNA (ssDNA). To avoid genome instability, RAD51 filaments are tightly controlled by a variety of positive and negative regulators. RADX (RPA-related RAD51-antagonist on the X chromosome) is a recently discovered negative regulator that binds tightly to ssDNA, directly interacts with RAD51, and regulates replication fork reversal and stabilization in a context-dependent manner. Here, we present a structure-based investigation of RADX's mechanism of action. Mass photometry experiments showed that RADX forms multiple oligomeric states in a concentration-dependent manner, with a predominance of trimers in the presence of ssDNA. The structure of RADX, which has no structurally characterized orthologs, was determined ab initio by cryo-electron microscopy (cryo-EM) from maps in the 2 to 4 Å range. The structure reveals the molecular basis for RADX oligomerization and the coupled multi-valent binding of ssDNA binding. The interaction of RADX with RAD51 filaments was imaged by negative stain EM, which showed a RADX oligomer at the end of filaments. Based on these results, we propose a model in which RADX functions by capping and restricting the end of RAD51 filaments. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_41940.map.gz | 447 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-41940-v30.xmlemd-41940.xml | 18.2 KB 18.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_41940_fsc.xml | 16.9 KB | Display | FSC data file |
| Images |  emd_41940.png emd_41940.png | 74.8 KB | ||
| Filedesc metadata | emd-41940.cif.gz | 6.5 KB | ||
| Others | emd_41940_half_map_1.map.gzemd_41940_half_map_2.map.gz | 474.4 MB 474.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-41940ftp://ftp.pdbj.org/pub/emdb/structures/EMD-41940 http://ftp.pdbj.org/pub/emdb/structures/EMD-41940ftp://ftp.pdbj.org/pub/emdb/structures/EMD-41940 | HTTPS FTP |
-Related structure data
| Related structure data |  8u61MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_41940.map.gz / Format: CCP4 / Size: 512 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | RADX tetramer bound to ssDNA dT25 | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.818 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: half map A
| File | emd_41940_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map A | ||||||||||||
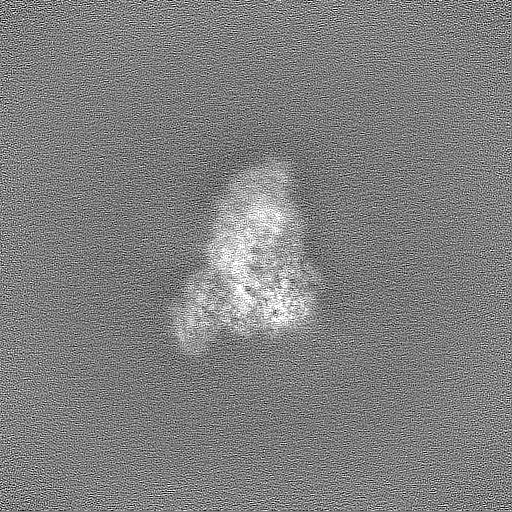
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: half map B
| File | emd_41940_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map B | ||||||||||||
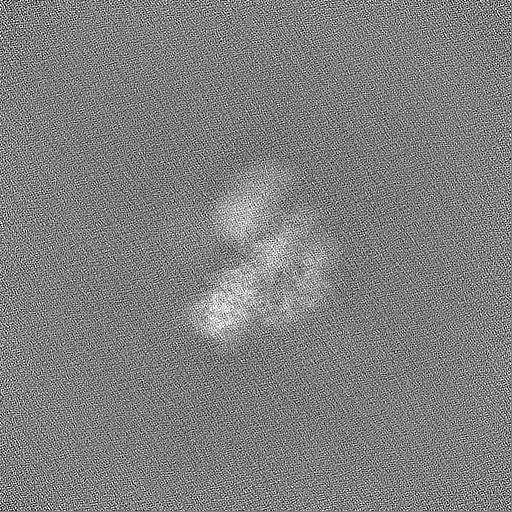
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : RADX tetramer bound to ssDNA dT25
| Entire | Name: RADX tetramer bound to ssDNA dT25 |
|---|---|
| Components |
|
-Supramolecule #1: RADX tetramer bound to ssDNA dT25
| Supramolecule | Name: RADX tetramer bound to ssDNA dT25 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 600 KDa |
-Macromolecule #1: RPA-related protein RADX
| Macromolecule | Name: RPA-related protein RADX / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 97.680094 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MSGESGQPEA GPSHAGLDWP NPERNRAGVP GGVIRRAGSQ GPRSWIQKVL EQIMDSPRQC VTPSEVVPVT VLAVQRYLLE DEPRDTVPK PPLYCYDVTI SDGVYQEKCY LDPSLNSLVY QNILKVGIQM RISRVSCLYN EKRIGQGILC IDNVHCGETS D SISLETPF ...String: MSGESGQPEA GPSHAGLDWP NPERNRAGVP GGVIRRAGSQ GPRSWIQKVL EQIMDSPRQC VTPSEVVPVT VLAVQRYLLE DEPRDTVPK PPLYCYDVTI SDGVYQEKCY LDPSLNSLVY QNILKVGIQM RISRVSCLYN EKRIGQGILC IDNVHCGETS D SISLETPF RNRAHQEKPE RPLRGGKSHY LALWNNEDPY GDIWLTDKQP EEHNFSDTKI ISLSHLEMTW TNRRNFPALL VR ILHKSKL RYYGKPDKKM IEPYQTFLEV ADSSGTVSVI MWNALCPEWY KSLRVGLVLL LQDYSVKKSY PFRIQPVPVD PQI KLISTM EICLNLRDPP TNIIIIPEKQ VKPEWRLPKL NHRFTTRSEL DDMPENCICD VIGLLVFVGR VQRSKKKENR EDFW SYRWI HIADGTSEQP FIVELFSTSQ PEIFENIYPM AYFVCTQLKV VRNDNQVPKL LYLTTTNESG VFITGHRGQP YTYDA KVKN FIQWIRTKSD SGEQKNMVIG GYYPYPPVPE TFSKYSSSIK VESLLTAISE VRKEIEDLQY REQKRIAIQG IITAIK YIP HSSATESASA SETLRNANRP STSQAARVEI QERNGKRHQD DEPVNSQYFQ TTSTNLSLSN KIRILQGPHA NPVAVPQ PG ASVQTKGIKP GMPSIFNRRA NINANLQGKA RKTISDRWES QLWREKKFGL IDHLHYSRVY PESIPRKFMF EHRKFLSD Q YNSQPAKYVP PEGRPPKLDD FKSARSLGHF EVTILGLNHE IAIDVAFLPM YCPEDIRTSQ IDTLLTSMNY SCAYPQDTT GNDRLPGPRA VAGDIIKAAT ELDRVHIVGI LDICNLGNNK VEVYLHKIYS PENTS UniProtKB: RPA-related protein RADX |
-Macromolecule #2: dT25 DNA (25-MER)
| Macromolecule | Name: dT25 DNA (25-MER) / type: dna / ID: 2 / Number of copies: 1 / Classification: DNA |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 7.559863 KDa |
| Sequence | String: (DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT) (DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT)(DT) (DT)(DT)(DT)(DT)(DT) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.145 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 20 sec. | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV | ||||||||||||||||||
| Details | Sample was cross-linked using BS3 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 1 / Average electron dose: 48.38 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.0 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-8u61: |
