Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-41817: Cryo-EM structure of the HSP90 dimer (NTD-MD) in the semi-open state -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of the HSP90 dimer (NTD-MD) in the semi-open state | |||||||||
 Map data Map data | Sharpened Map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | HSP / HSP90 / CHAPERONE | |||||||||
| Function / homology |  Function and homology information Function and homology informationprotein folding chaperone complex / ATP-dependent protein folding chaperone / : / ATP hydrolysis activity / ATP binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Trichoplusia ni (cabbage looper) Trichoplusia ni (cabbage looper) | |||||||||
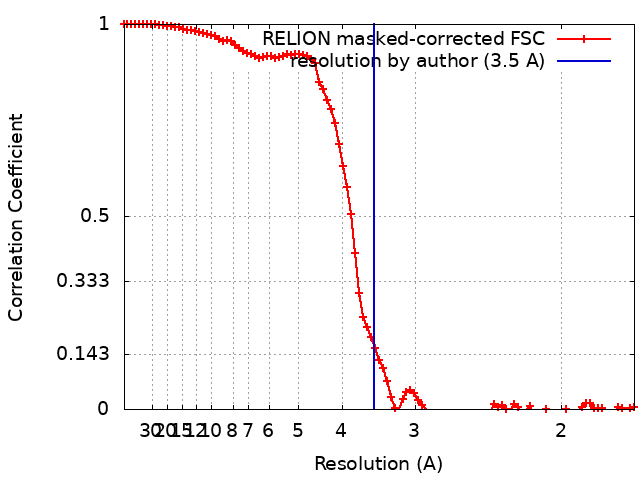
| Method | single particle reconstruction / cryo EM / Resolution: 3.5 Å | |||||||||
 Authors Authors | Finci LI / Simanshu DK | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Commun Biol / Year: 2024 Title: Structural dynamics of RAF1-HSP90-CDC37 and HSP90 complexes reveal asymmetric client interactions and key structural elements. Authors: Lorenzo I Finci / Mayukh Chakrabarti / Gulcin Gulten / Joseph Finney / Carissa Grose / Tara Fox / Renbin Yang / Dwight V Nissley / Frank McCormick / Dominic Esposito / Trent E Balius / Dhirendra K Simanshu / Abstract: RAF kinases are integral to the RAS-MAPK signaling pathway, and proper RAF1 folding relies on its interaction with the chaperone HSP90 and the cochaperone CDC37. Understanding the intricate molecular ...RAF kinases are integral to the RAS-MAPK signaling pathway, and proper RAF1 folding relies on its interaction with the chaperone HSP90 and the cochaperone CDC37. Understanding the intricate molecular interactions governing RAF1 folding is crucial for comprehending this process. Here, we present a cryo-EM structure of the closed-state RAF1-HSP90-CDC37 complex, where the C-lobe of the RAF1 kinase domain binds to one side of the HSP90 dimer, and an unfolded N-lobe segment of the RAF1 kinase domain threads through the center of the HSP90 dimer. CDC37 binds to the kinase C-lobe, mimicking the N-lobe with its HxNI motif. We also describe structures of HSP90 dimers without RAF1 and CDC37, displaying only N-terminal and middle domains, which we term the semi-open state. Employing 1 μs atomistic simulations, energetic decomposition, and comparative structural analysis, we elucidate the dynamics and interactions within these complexes. Our quantitative analysis reveals that CDC37 bridges the HSP90-RAF1 interaction, RAF1 binds HSP90 asymmetrically, and that HSP90 structural elements engage RAF1's unfolded region. Additionally, N- and C-terminal interactions stabilize HSP90 dimers, and molecular interactions in HSP90 dimers rearrange between the closed and semi-open states. Our findings provide valuable insight into the contributions of HSP90 and CDC37 in mediating client folding. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_41817.map.gz | 31.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-41817-v30.xmlemd-41817.xml | 24.2 KB 24.2 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_41817_fsc.xml | 9.2 KB | Display | FSC data file |
| Images | 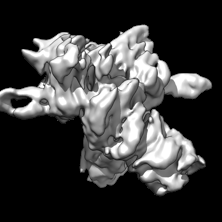 emd_41817.png emd_41817.png | 31.4 KB | ||
| Filedesc metadata | emd-41817.cif.gz | 6.6 KB | ||
| Others | emd_41817_additional_1.map.gzemd_41817_half_map_1.map.gzemd_41817_half_map_2.map.gz | 49.2 MB 49.6 MB 49.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-41817ftp://ftp.pdbj.org/pub/emdb/structures/EMD-41817 http://ftp.pdbj.org/pub/emdb/structures/EMD-41817ftp://ftp.pdbj.org/pub/emdb/structures/EMD-41817 | HTTPS FTP |
-Related structure data
| Related structure data |  8u1mMC  8u1lC  8u1nC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_41817.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Sharpened Map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.858 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Unsharpened Map
| File | emd_41817_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened Map | ||||||||||||
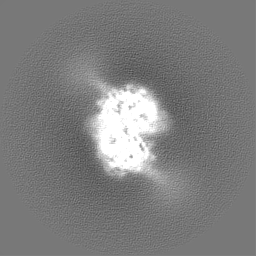
| Projections & Slices |
| ||||||||||||
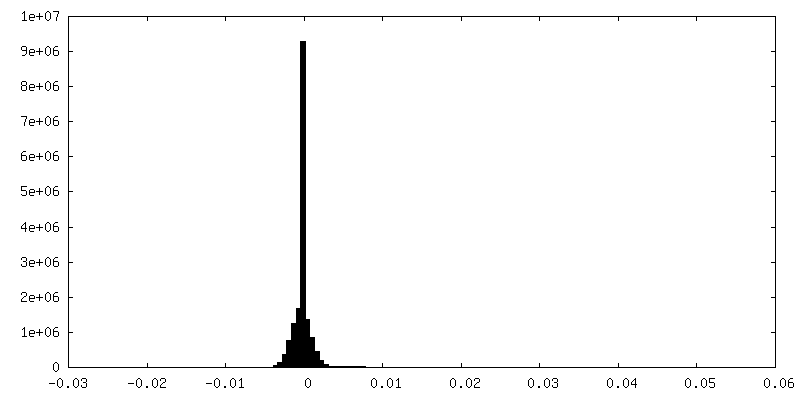
| Density Histograms |




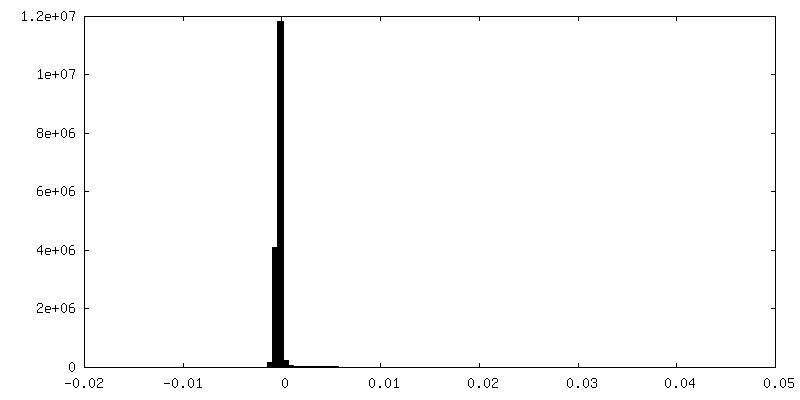
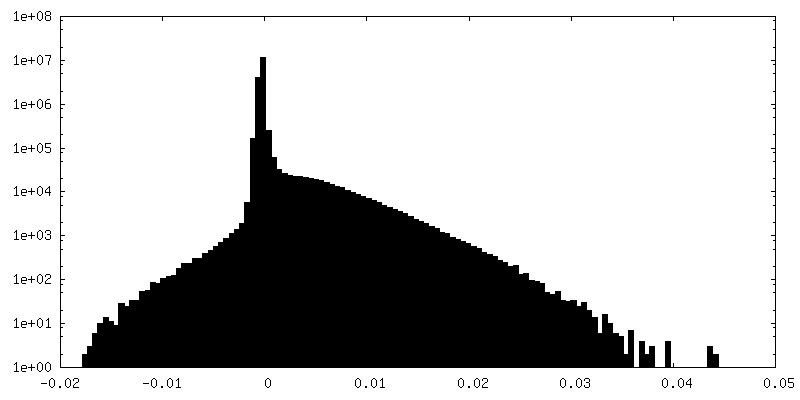
-Half map: Unfiltered, unsharpened half map 1
| File | emd_41817_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered, unsharpened half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




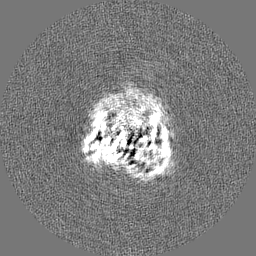


-Half map: Unfiltered, unsharpened half map 2
| File | emd_41817_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered, unsharpened half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Heat shock protein 90
| Entire | Name: Heat shock protein 90 |
|---|---|
| Components |
|
-Supramolecule #1: Heat shock protein 90
| Supramolecule | Name: Heat shock protein 90 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Trichoplusia ni (cabbage looper) |
| Molecular weight | Theoretical: 166 KDa |
-Macromolecule #1: Heat shock protein 83
| Macromolecule | Name: Heat shock protein 83 / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Trichoplusia ni (cabbage looper) |
| Molecular weight | Theoretical: 83.322133 KDa |
| Sequence | String: MPEEMQTDSG EVETFAFQAE IAQLMSLIIN TFYSNKEIFL RELISNSSDA LDKIRYESLT DPSKLDSGKE LYIKIIPNKS EGTFTIIDT GIGMTKADLV NNLGTIAKSG TKAFMEALQA GADISMIGQF GVGFYSCYLV ADRVTVHSKH NDDEQYMWES S AGGSFTVR ...String: MPEEMQTDSG EVETFAFQAE IAQLMSLIIN TFYSNKEIFL RELISNSSDA LDKIRYESLT DPSKLDSGKE LYIKIIPNKS EGTFTIIDT GIGMTKADLV NNLGTIAKSG TKAFMEALQA GADISMIGQF GVGFYSCYLV ADRVTVHSKH NDDEQYMWES S AGGSFTVR TDHGEPLGRG TKIVLHIKED LAEYLEVNKI KEIVKKHSQF IGYPIKLTVE KEREKELAYD EEEEKKEGEE DK KEDEKED EKPKIEDVGE DDEEDKDKKK KKTIKEKYTE DEELNKTKPI WTRNADDITQ EEYGDFYKSL TNDWEDHLAV KHF SVEGQL EFRALLFVPR RAPFDLFENK KRKNNIKLYV RRVFIMDNCE DLIPEYLNFI KGVVDSEDLP LNISREMLQQ NKIL KVIRK NLVKKCLELF EELAEDKENY KKYYEQFSKN LKLGIHEDAQ NRTKLADLLR YHTSASGDEA CSLKEYVSRM KENQK HIYY ITGENRDQVA NSSFVERVKK RGYEVVYMTE PIDEYVVQQM REYDGKTLVS VTKEGLELPE DEEEKKKREE DKVKFE GLC KVMKNILDNK VEKVVVSNRL VESPCCIVTA QYGWSANMER IMKAQALRDT STMGYMAAKK HLEINPDHSI VETLRQK AE ADKNDKAVKD LVILLYETAL LSSGFTLDEP QVHASRIYRM IKLGLGIDED EPIQVEESSV GDVPPLEGDA DDASRMEE V D UniProtKB: Heat shock protein 83 |
-Macromolecule #2: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 2 / Number of copies: 2 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Macromolecule #3: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 3 / Number of copies: 2 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.3 Component:
| |||||||||||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR | |||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV Details: Grids were blotted for 4.5 seconds before plunging. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.5 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |