 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-40852: Structure of the C-terminal protease CtpA-LbcA complex of Pseudom... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the C-terminal protease CtpA-LbcA complex of Pseudomonas aeruginosa | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | CtpA / LbcA / C-terminal protease / Pseudomonas aeruginosa / HYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationC-terminal processing peptidase / outer membrane-bounded periplasmic space / serine-type endopeptidase activity / signal transduction / proteolysis Similarity search - Function | |||||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) / Pseudomonas aeruginosa (bacteria) / | |||||||||
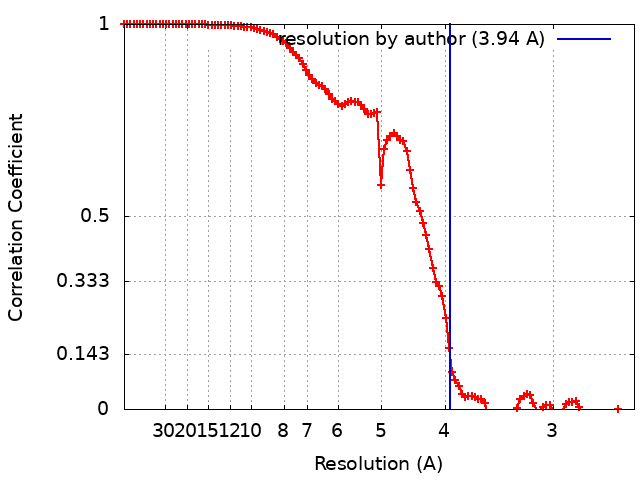
| Method | single particle reconstruction / cryo EM / Resolution: 3.94 Å | |||||||||
 Authors Authors | Hsu H-C / Li H | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: EMBO J / Year: 2024 Title: P. aeruginosa CtpA protease adopts a novel activation mechanism to initiate the proteolytic process. Authors: Hao-Chi Hsu / Michelle Wang / Amanda Kovach / Andrew J Darwin / Huilin Li / Abstract: During bacterial cell growth, hydrolases cleave peptide cross-links between strands of the peptidoglycan sacculus to allow new strand insertion. The Pseudomonas aeruginosa carboxyl-terminal ...During bacterial cell growth, hydrolases cleave peptide cross-links between strands of the peptidoglycan sacculus to allow new strand insertion. The Pseudomonas aeruginosa carboxyl-terminal processing protease (CTP) CtpA regulates some of these hydrolases by degrading them. CtpA assembles as an inactive hexamer composed of a trimer-of-dimers, but its lipoprotein binding partner LbcA activates CtpA by an unknown mechanism. Here, we report the cryo-EM structures of the CtpA-LbcA complex. LbcA has an N-terminal adaptor domain that binds to CtpA, and a C-terminal superhelical tetratricopeptide repeat domain. One LbcA molecule attaches to each of the three vertices of a CtpA hexamer. LbcA triggers relocation of the CtpA PDZ domain, remodeling of the substrate binding pocket, and realignment of the catalytic residues. Surprisingly, only one CtpA molecule in a CtpA dimer is activated upon LbcA binding. Also, a long loop from one CtpA dimer inserts into a neighboring dimer to facilitate the proteolytic activity. This work has revealed an activation mechanism for a bacterial CTP that is strikingly different from other CTPs that have been characterized structurally. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
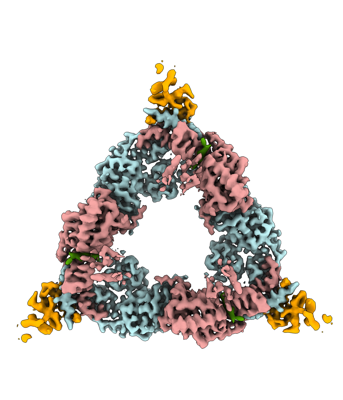
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_40852.map.gz | 118 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-40852-v30.xmlemd-40852.xml | 21.7 KB 21.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_40852_fsc.xml | 11.1 KB | Display | FSC data file |
| Images |  emd_40852.png emd_40852.png | 97.7 KB | ||
| Masks | emd_40852_msk_1.map | 125 MB | Mask map | |
| Filedesc metadata | emd-40852.cif.gz | 6.8 KB | ||
| Others | emd_40852_half_map_1.map.gzemd_40852_half_map_2.map.gz | 116 MB 116 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-40852ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40852 http://ftp.pdbj.org/pub/emdb/structures/EMD-40852ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40852 | HTTPS FTP |
-Related structure data
| Related structure data |  8sxhMC  8sxeC  8sxfC  8sxgC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



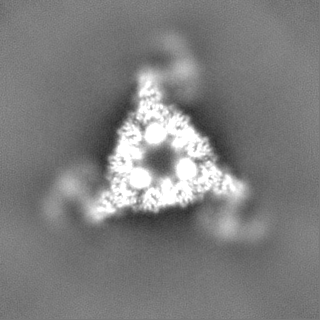
-Map
| File | Download / File: emd_40852.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.242 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_40852_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: #1
| File | emd_40852_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |


-Half map: #2
| File | emd_40852_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |

- Sample components
Sample components
-Entire : CtpA-LbcA complex of Pseudomonas aeruginosa
| Entire | Name: CtpA-LbcA complex of Pseudomonas aeruginosa |
|---|---|
| Components |
|
-Supramolecule #1: CtpA-LbcA complex of Pseudomonas aeruginosa
| Supramolecule | Name: CtpA-LbcA complex of Pseudomonas aeruginosa / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Pseudomonas aeruginosa (bacteria) |
| Molecular weight | Theoretical: 420 KDa |
-Macromolecule #1: Carboxyl-terminal protease
| Macromolecule | Name: Carboxyl-terminal protease / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Pseudomonas aeruginosa (bacteria) |
| Molecular weight | Theoretical: 42.920438 KDa |
| Recombinant expression | Organism: |
| Sequence | String: GSHMSAPLPL DELRTFAEVL DRVKAAYVEP VDDKTLLENA IKGMLSNLDP HSAYLGPEDF AELQESTSGE FGGLGIEVGS EDGFIKVVS PIDDTPAARA GIQPGDLIVQ IDGKPTKGQS MTEAVDSMRG KAGSPITLTI VRDGGRPFDV ELKRAIIKVK S VKSQVLEP ...String: GSHMSAPLPL DELRTFAEVL DRVKAAYVEP VDDKTLLENA IKGMLSNLDP HSAYLGPEDF AELQESTSGE FGGLGIEVGS EDGFIKVVS PIDDTPAARA GIQPGDLIVQ IDGKPTKGQS MTEAVDSMRG KAGSPITLTI VRDGGRPFDV ELKRAIIKVK S VKSQVLEP GYAYLRITQF QVNTGEEVVK ALNQLRKDNK GRLKGLVLDL RNNPGGVLQS AVEVADAFLT KGLIVYTKGR IA NSELRFS ADPADPSDKV PLVVLINGGS AAAAEIVAGA LQDQKRAILM GTDSFGKGSV QTVLPLNNDR ALKLTTALYY TPN GRSIQA QGIVPDIEVG RAKVTQERSS FEGFKEADLQ GHLANGNGGA DRPTGKRAAP SERPQDSDYQ LSQALSLLKG LSVT RGN UniProtKB: Carboxy-terminal processing protease CtpB |
-Macromolecule #2: TPR repeat-containing protein PA4667
| Macromolecule | Name: TPR repeat-containing protein PA4667 / type: protein_or_peptide / ID: 2 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Pseudomonas aeruginosa (bacteria) |
| Molecular weight | Theoretical: 61.742066 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MEDTAVETKA KPEKYGSFSE DSLYSLLVAE LAGQRNRFDI ALSNYVVQAQ KTRDPGVSER AFRIAEYLGA DQEALDTSLL WARSAPDNL DAQRAAAIQL ARAGRYEESM VYMEKVLNGQ GDTHFDFLAL SAAETDPDTR AGLLQSFDHL LKKYPNNGQL L FGKALLLQ ...String: MEDTAVETKA KPEKYGSFSE DSLYSLLVAE LAGQRNRFDI ALSNYVVQAQ KTRDPGVSER AFRIAEYLGA DQEALDTSLL WARSAPDNL DAQRAAAIQL ARAGRYEESM VYMEKVLNGQ GDTHFDFLAL SAAETDPDTR AGLLQSFDHL LKKYPNNGQL L FGKALLLQ QDGRPDEALT LLEDNSASRH EVAPLLLRSR LLQSMKRSDE ALPLLKAGIK EHPDDKRVRL AYARLLVEQN RL DDAKAEF AGLVQQFPDD DDLRFSLALV CLEAQAWDEA RIYLEELVER DSHVDAAHFN LGRLAEEQKD TARALDEYAQ VGP GNDFLP AQLRQTDVLL KAGRVDEAAQ RLDKARSEQP DYAIQLYLIE AEALSNNDQQ EKAWQAIQEG LKQYPEDLNL LYTR SMLAE KRNDLAQMEK DLRFVIAREP DNAMALNALG YTLADRTTRY GEARELILKA HKLNPDDPAI LDSMGWINYR QGKLA DAER YLRQALQRYP DHEVAAHLGE VLWAQGRQGD ARAIWREYLD KQPDSDVLRR TIKRLTGAET P UniProtKB: TPR repeat-containing protein PA4667 |
-Macromolecule #3: unidentified peptide
| Macromolecule | Name: unidentified peptide / type: protein_or_peptide / ID: 3 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 613.749 Da |
| Recombinant expression | Organism: |
| Sequence | String: (UNK)(UNK)(UNK)(UNK)(UNK)(UNK)(UNK) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.8 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: OTHER | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 279 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 11520 pixel / Digitization - Dimensions - Height: 8184 pixel / Number grids imaged: 1 / Number real images: 26257 / Average exposure time: 1.5 sec. / Average electron dose: 66.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.8 µm / Nominal defocus min: 1.3 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model |
| ||||||
|---|---|---|---|---|---|---|---|
| Refinement | Space: REAL / Protocol: AB INITIO MODEL / Overall B value: 196.3 | ||||||
| Output model | PDB-8sxh: |

