Journal: J Mol Biol / Year: 2001 Title: Crystal structures of the maltodextrin/maltose-binding protein complexed with reduced oligosaccharides: flexibility of tertiary structure and ligand binding. Authors: X Duan / J A Hall / H Nikaido / F A Quiocho / Abstract: The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a ...The structure of the maltodextrin or maltose-binding protein, an initial receptor for bacterial ABC-type active transport and chemotaxis, consists of two globular domains that are separated by a groove wherein the ligand is bound and enclosed by an inter-domain rotation. Here, we report the determination of the crystal structures of the protein complexed with reduced maltooligosaccharides (maltotriitol and maltotetraitol) in both the "closed" and "open" forms. Although these modified sugars bind to the receptor, they are not transported by the wild-type transporter. In the closed structures, the reduced sugars are buried in the groove and bound by both domains, one domain mainly by hydrogen-bonding interactions and the other domain primarily by non-polar interactions with aromatic side-chains. In the open structures, which abrogate both cellular activities of active transport and chemotaxis because of the large separation between the two domains, the sugars are bound almost exclusively to the domain rich in aromatic residues. The binding site for the open chain glucitol residue extends to a subsite that is distinct from those for the glucose residues that were uncovered in prior structural studies of the binding of active linear maltooligosaccharides. Occupation of this subsite may also account for the inability of the reduced oligosaccharides to be transported. The structures reported here, combined with those previously determined for several other complexes with active oligosaccharides in the closed form and with cyclodextrin in the open form, revealed at least four distinct modes of ligand binding but with only one being functionally active. This versatility reflects the flexibility of the protein, from very large motions of interdomain rotation to more localized side-chain conformational changes, and adaptation by the oligosaccharides as well.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors Citation
Citation
 Structure visualization
Structure visualization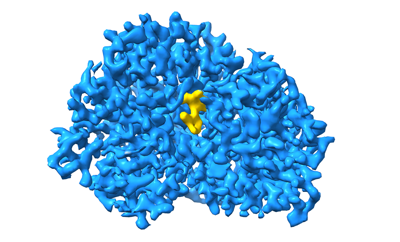
 Downloads & links
Downloads & links emd_39117.png
emd_39117.png http://ftp.pdbj.org/pub/emdb/structures/EMD-39117
http://ftp.pdbj.org/pub/emdb/structures/EMD-39117



 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















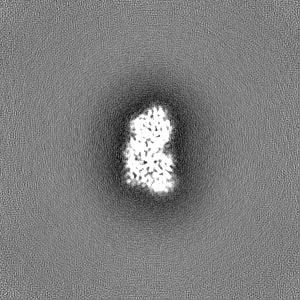
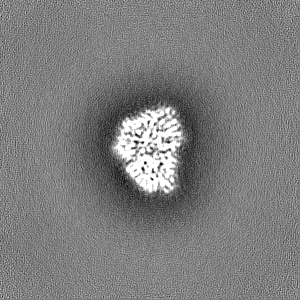
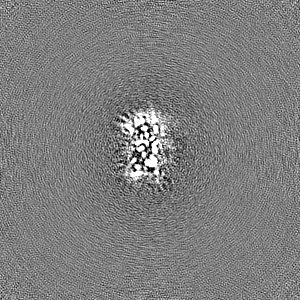
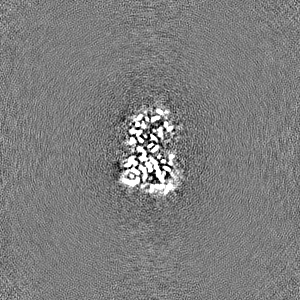
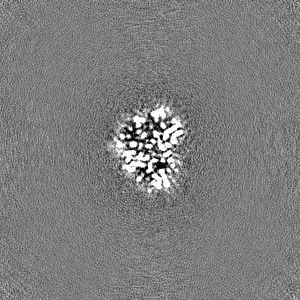
 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN