 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of native H. thermoluteolus TH-1 GroEL | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Chaperone / Chaperonin / ATPase / protein folding | |||||||||
| Function / homology |  Function and homology information Function and homology informationchaperonin ATPase / isomerase activity / ATP-dependent protein folding chaperone / : / protein refolding / ATP binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  Hydrogenophilus thermoluteolus (bacteria) Hydrogenophilus thermoluteolus (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.3 Å | |||||||||
 Authors Authors | Liao Z / Gopalasingam CC / Kameya M / Gerle C / Shigematsu H / Ishii M / Arakawa T / Fushinobu S | |||||||||
| Funding support |  Japan, 2 items Japan, 2 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2024 Title: Structural insights into thermophilic chaperonin complexes. Authors: Zengwei Liao / Chai C Gopalasingam / Masafumi Kameya / Christoph Gerle / Hideki Shigematsu / Masaharu Ishii / Takatoshi Arakawa / Shinya Fushinobu / Abstract: Group I chaperonins are dual heptamer protein complexes that play significant roles in protein homeostasis. The structure and function of the Escherichia coli chaperonin are well characterized. ...Group I chaperonins are dual heptamer protein complexes that play significant roles in protein homeostasis. The structure and function of the Escherichia coli chaperonin are well characterized. However, the dynamic properties of chaperonins, such as large ATPase-dependent conformational changes by binding of lid-like co-chaperonin GroES, have made structural analyses challenging, and our understanding of these changes during the turnover of chaperonin complex formation is limited. In this study, we used single-particle cryogenic electron microscopy to investigate the structures of GroES-bound chaperonin complexes from the thermophilic hydrogen-oxidizing bacteria Hydrogenophilus thermoluteolus and Hydrogenobacter thermophilus in the presence of ATP and AMP-PNP. We captured the structure of an intermediate state chaperonin complex, designated as an asymmetric football-shaped complex, and performed analyses to decipher the dynamic structural variations. Our structural analyses of inter- and intra-subunit communications revealed a unique mechanism of complex formation through the binding of a second GroES to a bullet-shaped complex. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
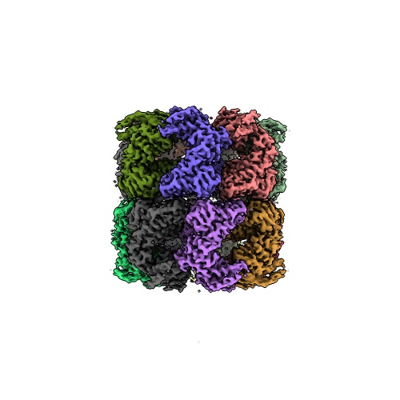
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_37850.map.gz | 88.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-37850-v30.xmlemd-37850.xml | 20.6 KB 20.6 KB | Display Display | EMDB header |
| Images |  emd_37850.png emd_37850.png | 83.4 KB | ||
| Masks | emd_37850_msk_1.map | 178 MB | Mask map | |
| Filedesc metadata | emd-37850.cif.gz | 6.2 KB | ||
| Others | emd_37850_additional_1.map.gzemd_37850_half_map_1.map.gzemd_37850_half_map_2.map.gz | 168.1 MB 165.2 MB 165.2 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-37850ftp://ftp.pdbj.org/pub/emdb/structures/EMD-37850 http://ftp.pdbj.org/pub/emdb/structures/EMD-37850ftp://ftp.pdbj.org/pub/emdb/structures/EMD-37850 | HTTPS FTP |
-Related structure data
| Related structure data |  8wu4MC  8wucC  8wuwC  8wuxC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_37850.map.gz / Format: CCP4 / Size: 178 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
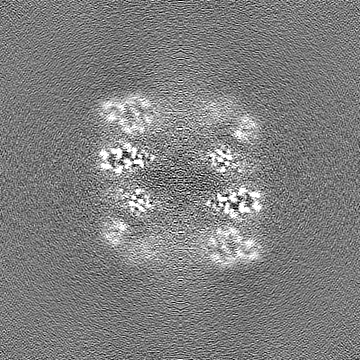
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.84 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_37850_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






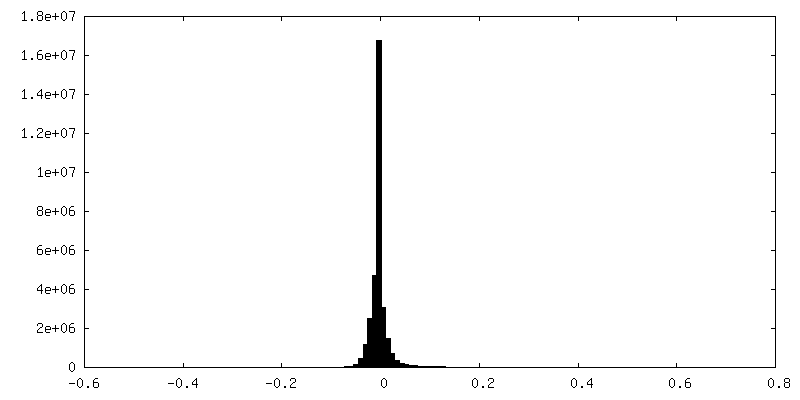
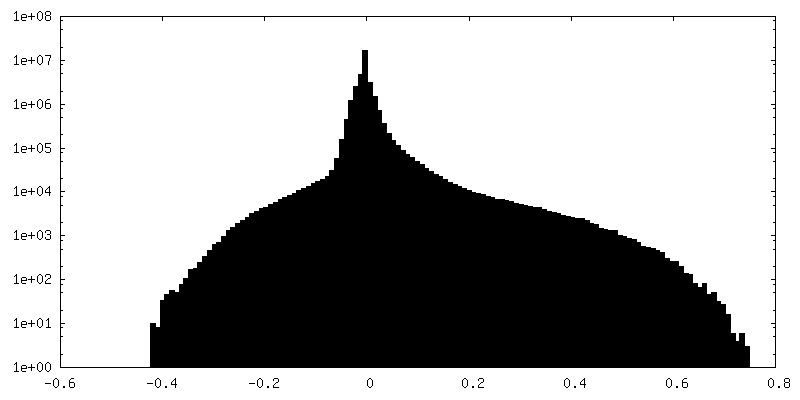
-Additional map: Output sharpened map from NU-refinement of cryoSPARC
| File | emd_37850_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Output sharpened map from NU-refinement of cryoSPARC | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




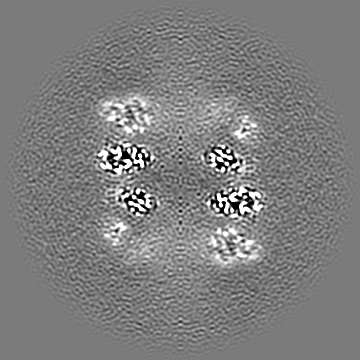

-Half map: #1
| File | emd_37850_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |



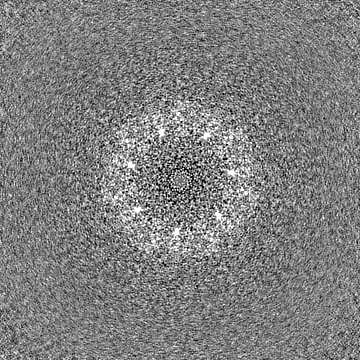
-Half map: #2
| File | emd_37850_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |

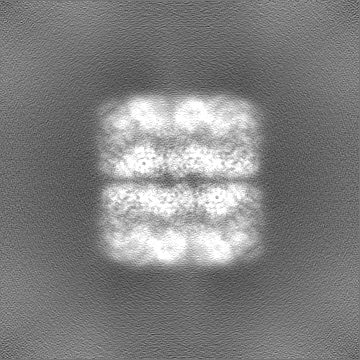





- Sample components
Sample components
-Entire : Native H. thermoluteolus GroEL
| Entire | Name: Native H. thermoluteolus GroEL |
|---|---|
| Components |
|
-Supramolecule #1: Native H. thermoluteolus GroEL
| Supramolecule | Name: Native H. thermoluteolus GroEL / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Hydrogenophilus thermoluteolus (bacteria) / Strain: TH-1 |
| Molecular weight | Theoretical: 809 kDa/nm |
-Macromolecule #1: Chaperonin GroEL
| Macromolecule | Name: Chaperonin GroEL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO / EC number: chaperonin ATPase |
|---|---|
| Source (natural) | Organism: Hydrogenophilus thermoluteolus (bacteria) / Strain: TH-1 |
| Molecular weight | Theoretical: 55.83707 KDa |
| Sequence | String: AAKEVKFHDS ARERLVAGVN LLANAVKTTL GPKGRNVVIE RSFGAPIVTK DGVTVAKEIE LKDKFENMGA QMVKEVASKT ADVAGDGTT TATVLAQAIV REGMKYVAAG MNPMDLKRGI DKAVTAIVEE LKAISKPCST TKEIAQVGTI SANADSSIGE I IAQAMDKV ...String: AAKEVKFHDS ARERLVAGVN LLANAVKTTL GPKGRNVVIE RSFGAPIVTK DGVTVAKEIE LKDKFENMGA QMVKEVASKT ADVAGDGTT TATVLAQAIV REGMKYVAAG MNPMDLKRGI DKAVTAIVEE LKAISKPCST TKEIAQVGTI SANADSSIGE I IAQAMDKV GKEGVITVED GKSLENELEV VEGMQFDRGY LSPYFINNPD KQVAVLDNPY ILLHDKKISN IRDLLPVLEQ VA KAGRPLL IIAEDVEGEA LATLVVNNLR GILKTCAVKA PGFGDRRKAM LQDIAILTGG TVISEEVGLS LEKATLEDLG QAK RVEVAK EHTTIIDGAG DPAKIQARVK EIRVQIEEAT SDYDREKLQE RVAKLAGGVA VIKVGAATEV EMKEKKARVE DALH ATRAA VEEGIVPGGG VALLRAREAA VAKGLKGDNP DQEAGIKIVL RAVEQPLREI VANAGEEPSV IVAKVLEGKG NYGYN AATG EFGDMIEMGV LDPTKVTRSA LQNAASVAGL MLTTECMIAE AP UniProtKB: Chaperonin GroEL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
| ||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 90 sec. / Pretreatment - Atmosphere: OTHER / Details: PIB-10, hard mode | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV / Details: blot force 10, blot time 15 s. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: FEI FALCON IV (4k x 4k) / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 3418 / Average exposure time: 4.95 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 120000 |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | Chain - Source name: AlphaFold / Chain - Initial model type: in silico model |
|---|---|
| Details | Initial fitting was done using UCSF ChimeraX |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-8wu4: |
