National Institutes of Health/National Institute Of Allergy and Infectious Diseases (NIH/NIAID)
U54 AI170855
United States
National Institutes of Health/National Institute Of Allergy and Infectious Diseases (NIH/NIAID)
U01 AI136680
United States
National Institutes of Health/Office of the Director
GM148476
United States
National Institutes of Health/Office of the Director
GM082946
United States
National Science Foundation (NSF, United States)
MCB 2048095
United States
Other private
Hearst Foundations Developmental Chair
Other private
Margaret T. Morris Foundation
Other government
National University of Singapore (A-0008405-00-00, A-0008405-01-00)
Ministry of Education (MoE, Singapore)
A-8000037-00-00
Singapore
National Research Foundation (NRF, Singapore)
A-8001346-00-00
Singapore
Other government
Agency for Science, Technology and Research Singapore
Damon Runyon Cancer Research Foundation
DRR-65-21
United States
American Cancer Society
PF-21-075-01-CCB
United States
National Institutes of Health/Office of the Director
R01-GM129325
United States
National Institutes of Health/Office of the Director
GM103310
United States
Simons Foundation
SF349247
United States
Other government
NYSTAR
National Institutes of Health/Office of the Director
S10 OD032467
United States
Citation
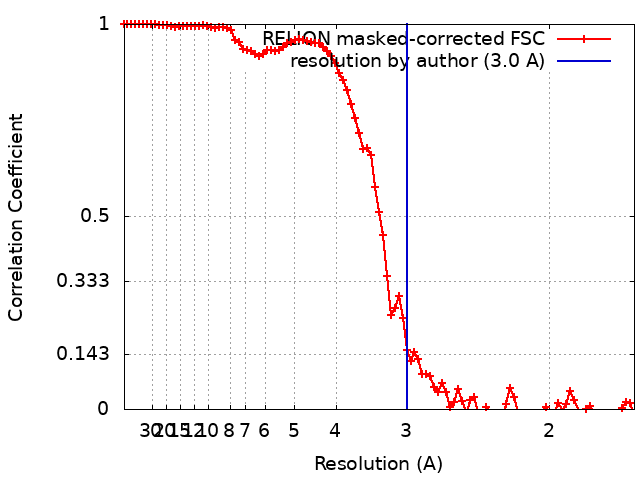
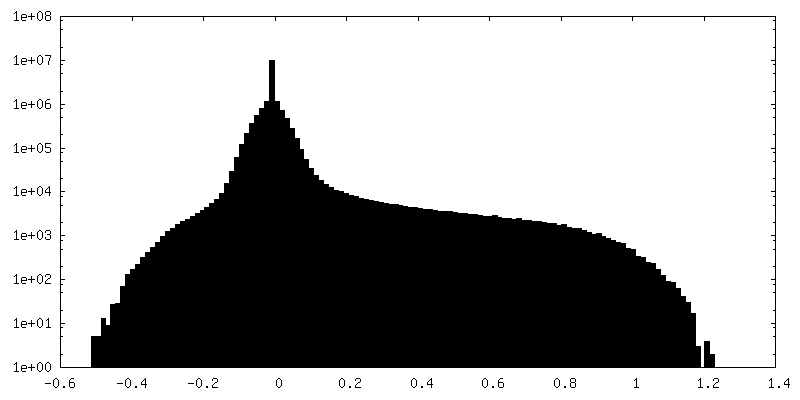
Journal: Nat Commun / Year: 2024 Title: Overcoming resolution attenuation during tilted cryo-EM data collection. Authors: Sriram Aiyer / Philip R Baldwin / Shi Min Tan / Zelin Shan / Juntaek Oh / Atousa Mehrani / Marianne E Bowman / Gordon Louie / Dario Oliveira Passos / Selena Đorđević-Marquardt / Mario ...Authors: Sriram Aiyer / Philip R Baldwin / Shi Min Tan / Zelin Shan / Juntaek Oh / Atousa Mehrani / Marianne E Bowman / Gordon Louie / Dario Oliveira Passos / Selena Đorđević-Marquardt / Mario Mietzsch / Joshua A Hull / Shuichi Hoshika / Benjamin A Barad / Danielle A Grotjahn / Robert McKenna / Mavis Agbandje-McKenna / Steven A Benner / Joseph A P Noel / Dong Wang / Yong Zi Tan / Dmitry Lyumkis / Abstract: Structural biology efforts using cryogenic electron microscopy are frequently stifled by specimens adopting "preferred orientations" on grids, leading to anisotropic map resolution and impeding ...Structural biology efforts using cryogenic electron microscopy are frequently stifled by specimens adopting "preferred orientations" on grids, leading to anisotropic map resolution and impeding structure determination. Tilting the specimen stage during data collection is a generalizable solution but has historically led to substantial resolution attenuation. Here, we develop updated data collection and image processing workflows and demonstrate, using multiple specimens, that resolution attenuation is negligible or significantly reduced across tilt angles. Reconstructions with and without the stage tilted as high as 60° are virtually indistinguishable. These strategies allowed the reconstruction to 3 Å resolution of a bacterial RNA polymerase with preferred orientation, containing an unnatural nucleotide for studying novel base pair recognition. Furthermore, we present a quantitative framework that allows cryo-EM practitioners to define an optimal tilt angle during data acquisition. These results reinforce the utility of employing stage tilt for data collection and provide quantitative metrics to obtain isotropic maps.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords
 Authors
Authors United States,
United States,  Singapore, 18 items
Singapore, 18 items  Citation
Citation
 Structure visualization
Structure visualization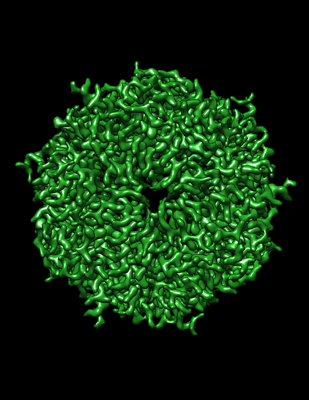
 Downloads & links
Downloads & links EMDB map data format
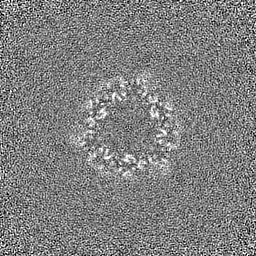
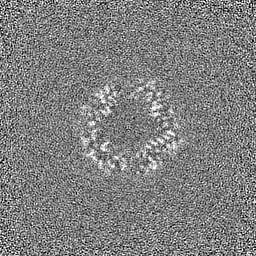
EMDB map data format emd_36819.png
emd_36819.png http://ftp.pdbj.org/pub/emdb/structures/EMD-36819
http://ftp.pdbj.org/pub/emdb/structures/EMD-36819























 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


































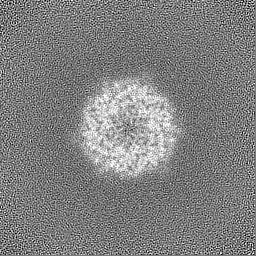
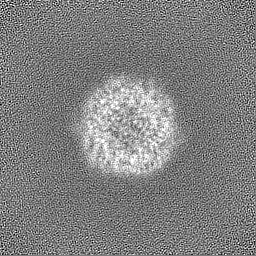


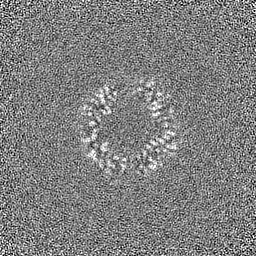
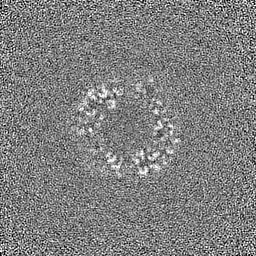


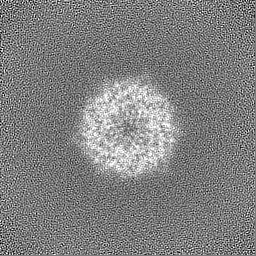
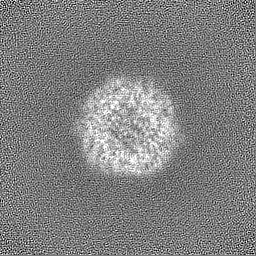
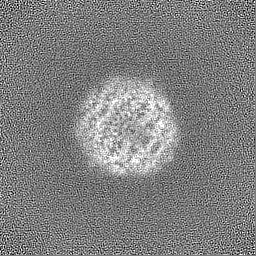


 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN