 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of human TMEM87A, gluconate-bound | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | non-selective cation channel / ion channel / membrane protein / Golgi-localized protein | |||||||||
| Function / homology |  Function and homology information Function and homology informationdetection of mechanical stimulus involved in sensory perception of touch / retrograde transport, endosome to Golgi / Golgi cisterna membrane / RHOA GTPase cycle / ruffle / bioluminescence / generation of precursor metabolites and energy / cellular response to mechanical stimulus / Golgi membrane / Golgi apparatus ...detection of mechanical stimulus involved in sensory perception of touch / retrograde transport, endosome to Golgi / Golgi cisterna membrane / RHOA GTPase cycle / ruffle / bioluminescence / generation of precursor metabolites and energy / cellular response to mechanical stimulus / Golgi membrane / Golgi apparatus / plasma membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) / Homo sapiens (human) /   Human adenovirus 2 Human adenovirus 2 | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.6 Å | |||||||||
 Authors Authors | Han A / Kim HM | |||||||||
| Funding support |  Korea, Republic Of, 1 items Korea, Republic Of, 1 items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2024 Title: GolpHCat (TMEM87A), a unique voltage-dependent cation channel in Golgi apparatus, contributes to Golgi-pH maintenance and hippocampus-dependent memory. Authors: Hyunji Kang / Ah-Reum Han / Aihua Zhang / Heejin Jeong / Wuhyun Koh / Jung Moo Lee / Hayeon Lee / Hee Young Jo / Miguel A Maria-Solano / Mridula Bhalla / Jea Kwon / Woo Suk Roh / Jimin Yang ...Authors: Hyunji Kang / Ah-Reum Han / Aihua Zhang / Heejin Jeong / Wuhyun Koh / Jung Moo Lee / Hayeon Lee / Hee Young Jo / Miguel A Maria-Solano / Mridula Bhalla / Jea Kwon / Woo Suk Roh / Jimin Yang / Hyun Joo An / Sun Choi / Ho Min Kim / C Justin Lee / Abstract: Impaired ion channels regulating Golgi pH lead to structural alterations in the Golgi apparatus, such as fragmentation, which is found, along with cognitive impairment, in Alzheimer's disease. ...Impaired ion channels regulating Golgi pH lead to structural alterations in the Golgi apparatus, such as fragmentation, which is found, along with cognitive impairment, in Alzheimer's disease. However, the causal relationship between altered Golgi structure and cognitive impairment remains elusive due to the lack of understanding of ion channels in the Golgi apparatus of brain cells. Here, we identify that a transmembrane protein TMEM87A, renamed Golgi-pH-regulating cation channel (GolpHCat), expressed in astrocytes and neurons that contributes to hippocampus-dependent memory. We find that GolpHCat displays unique voltage-dependent currents, which is potently inhibited by gluconate. Additionally, we gain structural insights into the ion conduction through GolpHCat at the molecular level by determining three high-resolution cryogenic-electron microscopy structures of human GolpHCat. GolpHCat-knockout mice show fragmented Golgi morphology and altered protein glycosylation and functions in the hippocampus, leading to impaired spatial memory. These findings suggest a molecular target for Golgi-related diseases and cognitive impairment. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_35017.map.gz | 62 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-35017-v30.xmlemd-35017.xml | 21.6 KB 21.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_35017_fsc.xml | 12 KB | Display | FSC data file |
| Images |  emd_35017.png emd_35017.png | 42.6 KB | ||
| Filedesc metadata | emd-35017.cif.gz | 7.6 KB | ||
| Others | emd_35017_half_map_1.map.gzemd_35017_half_map_2.map.gz | 116.1 MB 116.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-35017ftp://ftp.pdbj.org/pub/emdb/structures/EMD-35017 http://ftp.pdbj.org/pub/emdb/structures/EMD-35017ftp://ftp.pdbj.org/pub/emdb/structures/EMD-35017 | HTTPS FTP |
-Related structure data
| Related structure data |  8httMC  8hsiC  8kb4C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |


-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_35017.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.849 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: #2
| File | emd_35017_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_35017_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |




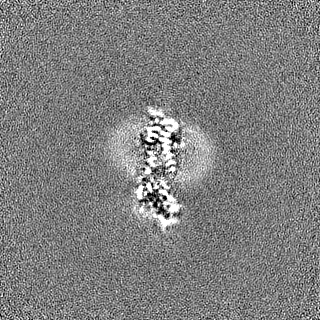



- Sample components
Sample components
-Entire : Transmembrane protein 87A with GFP tag and Twin-strep tag with gl...
| Entire | Name: Transmembrane protein 87A with GFP tag and Twin-strep tag with gluconate |
|---|---|
| Components |
|
-Supramolecule #1: Transmembrane protein 87A with GFP tag and Twin-strep tag with gl...
| Supramolecule | Name: Transmembrane protein 87A with GFP tag and Twin-strep tag with gluconate type: organelle_or_cellular_component / ID: 1 / Parent: 0 / Macromolecule list: #1 Details: Transmembrane protein 87A with GFP tag and Twin-strep tag with gluconate purified with detergent LMNG/CHS |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
-Macromolecule #1: Transmembrane protein 87A,EGFP
| Macromolecule | Name: Transmembrane protein 87A,EGFP / type: protein_or_peptide / ID: 1 Details: The chimera of Transmembrane protein 87A, Linkers, EGFP and Tags Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Human adenovirus 2 |
| Molecular weight | Theoretical: 97.965617 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MAAAAWLQVL PVILLLLGAH PSPLSFFSAG PATVAAADRS KWHIPIPSGK NYFSFGKILF RNTTIFLKFD GEPCDLSLNI TWYLKSADC YNEIYNFKAE EVELYLEKLK EKRGLSGKYQ TSSKLFQNCS ELFKTQTFSG DFMHRLPLLG EKQEAKENGT N LTFIGDKT ...String: MAAAAWLQVL PVILLLLGAH PSPLSFFSAG PATVAAADRS KWHIPIPSGK NYFSFGKILF RNTTIFLKFD GEPCDLSLNI TWYLKSADC YNEIYNFKAE EVELYLEKLK EKRGLSGKYQ TSSKLFQNCS ELFKTQTFSG DFMHRLPLLG EKQEAKENGT N LTFIGDKT AMHEPLQTWQ DAPYIFIVHI GISSSKESSK ENSLSNLFTM TVEVKGPYEY LTLEDYPLMI FFMVMCIVYV LF GVLWLAW SACYWRDLLR IQFWIGAVIF LGMLEKAVFY AEFQNIRYKG ESVQGALILA ELLSAVKRSL ARTLVIIVSL GYG IVKPRL GVTLHKVVVA GALYLLFSGM EGVLRVTGAQ TDLASLAFIP LAFLDTALCW WIFISLTQTM KLLKLRRNIV KLSL YRHFT NTLILAVAAS IVFIIWTTMK FRIVTCQSDW RELWVDDAIW RLLFSMILFV IMVLWRPSAN NQRFAFSPLS EEEEE DEQK EPMLKESFEG MKMRSTKQEP NGNSKVNKAQ EDDLKWVEEN VPSSVTDVAL PALLDSDEER MITHFERSKM ELKENL YFQ GGTLEVLFQG PNPAFLYKVV DPVVSKGEEL FTGVVPILVE LDGDVNGHKF SVSGEGEGDA TYGKLTLKFI CTTGKLP VP WPTLVTTLTY GVQCFSRYPD HMKQHDFFKS AMPEGYVQER TIFFKDDGNY KTRAEVKFEG DTLVNRIELK GIDFKEDG N ILGHKLEYNY NSHNVYIMAD KQKNGIKVNF KIRHNIEDGS VQLADHYQQN TPIGDGPVLL PDNHYLSTQS ALSKDPNEK RDHMVLLEFV TAAGITLGMD ELYKEFLVPR GSSRSAWSHP QFEKGGGSGG GSGGSAWSHP QFEK UniProtKB: Transmembrane protein 87A, EGFP |
-Macromolecule #4: D-gluconic acid
| Macromolecule | Name: D-gluconic acid / type: ligand / ID: 4 / Number of copies: 1 / Formula: GCO |
|---|---|
| Molecular weight | Theoretical: 196.155 Da |
| Chemical component information | 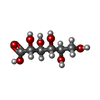 ChemComp-GCO: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.7 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 9 Component:
Details: 50mM HEPES pH 7.5, 250mM NaCl, 0.01% (w/v) LMNG, 0.002% (w/v) CHS | |||||||||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV | |||||||||||||||
| Details | This sample was monodisperse |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Alignment procedure | Coma free - Residual tilt: 10.0 mrad |
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Details | Using Zemlin tableau in sherpa |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 13099 / Average exposure time: 6.14 sec. / Average electron dose: 68.15 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Calibrated magnification: 58900 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 1.9000000000000001 µm / Nominal defocus min: 0.8 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Details | Chimera Fit in map tool was used for initial local fitting. Then, Real-space refinement with the rigid body option in PHENIX was used for flexible fitting. |
|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Overall B value: 40 |
| Output model | PDB-8htt: |
