 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-2983 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
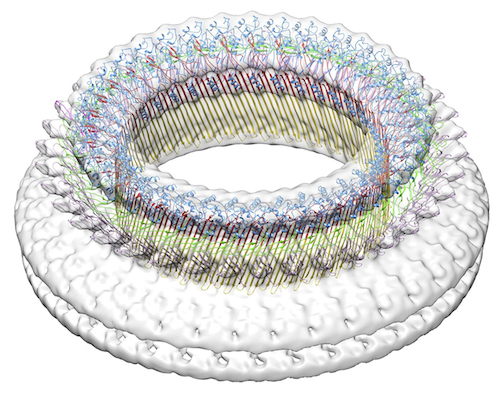
| タイトル | Cryo EM structure of suilysin pore | |||||||||
 マップデータ マップデータ | reconstruction of suilysin pore on liposome | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | cholesterol dependent cytolysin / hemolysin / toxin | |||||||||
| 機能・相同性 | :  機能・相同性情報 機能・相同性情報 | |||||||||
| 生物種 |  Streptococcus suis (バクテリア) Streptococcus suis (バクテリア) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 15.0 Å | |||||||||
 データ登録者 データ登録者 | Dudkina NV / Leung C / Lukoyanova N / Hodel AW / Farabella I / Pandurangan AP / Jahan N / Pires Damaso M / Osmanovic D / Reboul CF ...Dudkina NV / Leung C / Lukoyanova N / Hodel AW / Farabella I / Pandurangan AP / Jahan N / Pires Damaso M / Osmanovic D / Reboul CF / Dunstone MA / Andrew PW / Lonnen R / Topf M / Saibil HR / Hoogenboom BW | |||||||||
 引用 引用 | ジャーナル: Elife / 年: 2014 タイトル: Stepwise visualization of membrane pore formation by suilysin, a bacterial cholesterol-dependent cytolysin. 著者: Carl Leung / Natalya V Dudkina / Natalya Lukoyanova / Adrian W Hodel / Irene Farabella / Arun P Pandurangan / Nasrin Jahan / Mafalda Pires Damaso / Dino Osmanović / Cyril F Reboul / Michelle ...著者: Carl Leung / Natalya V Dudkina / Natalya Lukoyanova / Adrian W Hodel / Irene Farabella / Arun P Pandurangan / Nasrin Jahan / Mafalda Pires Damaso / Dino Osmanović / Cyril F Reboul / Michelle A Dunstone / Peter W Andrew / Rana Lonnen / Maya Topf / Helen R Saibil / Bart W Hoogenboom /   要旨: Membrane attack complex/perforin/cholesterol-dependent cytolysin (MACPF/CDC) proteins constitute a major superfamily of pore-forming proteins that act as bacterial virulence factors and effectors in ...Membrane attack complex/perforin/cholesterol-dependent cytolysin (MACPF/CDC) proteins constitute a major superfamily of pore-forming proteins that act as bacterial virulence factors and effectors in immune defence. Upon binding to the membrane, they convert from the soluble monomeric form to oligomeric, membrane-inserted pores. Using real-time atomic force microscopy (AFM), electron microscopy (EM), and atomic structure fitting, we have mapped the structure and assembly pathways of a bacterial CDC in unprecedented detail and accuracy, focussing on suilysin from Streptococcus suis. We show that suilysin assembly is a noncooperative process that is terminated before the protein inserts into the membrane. The resulting ring-shaped pores and kinetically trapped arc-shaped assemblies are all seen to perforate the membrane, as also visible by the ejection of its lipids. Membrane insertion requires a concerted conformational change of the monomeric subunits, with a marked expansion in pore diameter due to large changes in subunit structure and packing. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_2983.map.gz | 17.7 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-2983-v30.xmlemd-2983.xml | 11.3 KB 11.3 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  Slypore.jpg Slypore.jpg | 207.5 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2983 http://ftp.pdbj.org/pub/emdb/structures/EMD-2983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2983 | HTTPS FTP |
-関連構造データ



-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_2983.map.gz / 形式: CCP4 / 大きさ: 238.4 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | reconstruction of suilysin pore on liposome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : Suilysin
| 全体 | 名称: Suilysin |
|---|---|
| 要素 |
|
-超分子 #1000: Suilysin
| 超分子 | 名称: Suilysin / タイプ: sample / ID: 1000 詳細: The protein was incubated with lipid vesicles (PC:Cholesterol, molar ratio 45:55) for 10 minutes at 37oC 集合状態: 37 / Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 2.072 MDa / 理論値: 2.072 MDa / 手法: Calculated from the molecular weight of the monomer |
-分子 #1: suilysin
| 分子 | 名称: suilysin / タイプ: protein_or_peptide / ID: 1 / Name.synonym: SLY / コピー数: 1 / 集合状態: 37 / 組換発現: Yes |
|---|---|
| 由来(天然) | 生物種: Streptococcus suis (バクテリア) / 細胞: bacterium |
| 分子量 | 実験値: 56 KDa / 理論値: 56 KDa |
| 組換発現 | 生物種: |
| 配列 | UniProtKB: UNIPROTKB: C6GNG6 |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.5 mg/mL |
|---|---|
| 緩衝液 | pH: 7.5 / 詳細: 50 mM HEPES, 100 mM NaCl |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 91 K / 装置: FEI VITROBOT MARK III 手法: Liposomes with the protein were applied to glow-discharged lacey carbon coated copper grids and blotted for 5 s. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI POLARA 300 |
|---|---|
| 温度 | 最低: 80 K / 最高: 90 K / 平均: 85 K |
| アライメント法 | Legacy - 非点収差: Objective lens astigmatism was corrected at 150,000x magnification |
| 日付 | 2013年12月1日 |
| 撮影 | カテゴリ: CCD フィルム・検出器のモデル: GATAN ULTRASCAN 4000 (4k x 4k) デジタル化 - サンプリング間隔: 15 µm / 実像数: 7128 / 平均電子線量: 25 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 75000 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.3 mm / 最大 デフォーカス(公称値): 4.0 µm / 最小 デフォーカス(公称値): 1.5 µm / 倍率(公称値): 50000 |
| 試料ステージ | 試料ホルダーモデル: GATAN HELIUM |
| 実験機器 |  モデル: Tecnai Polara / 画像提供: FEI Company |
-画像解析
| 詳細 | 37-fold symmetrised reconstruction. Image regions containing the pores with small surrounding areas of membrane were selected manually using Boxer (EMAN 1.9) software. |
|---|---|
| CTF補正 | 詳細: Phase flipping for each particle |
| 最終 再構成 | 想定した対称性 - 点群: C37 (37回回転対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 15.0 Å / 解像度の算出法: OTHER / ソフトウェア - 名称: SPIDER, IMAGIC, EMAN / 詳細: Final maps were calculated by BP RP in SPIDER / 使用した粒子像数: 600 |
-原子モデル構築 1
| 初期モデル | PDB ID: Chain - Chain ID: A |
|---|---|
| ソフトウェア | 名称: Chimera |
| 詳細 | Rigid body fitting of domain models created as described in Leung et al (2014) eLife 3:e04247 |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT 当てはまり具合の基準: Cross-correlation coefficient |

