National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
GM131754
United States
Citation
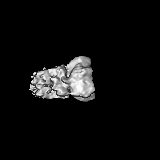
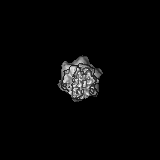
Journal: J Biol Chem / Year: 2023 Title: The Saccharomyces cerevisiae Yta7 ATPase hexamer contains a unique bromodomain tier that functions in nucleosome disassembly. Authors: Feng Wang / Xiang Feng / Qing He / Hua Li / Huilin Li / Abstract: The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to ...The Saccharomyces cerevisiae Yta7 is a chromatin remodeler harboring a histone-interacting bromodomain (BRD) and two AAA+ modules. It is not well understood how Yta7 recognizes the histone H3 tail to promote nucleosome disassembly for DNA replication or RNA transcription. By cryo-EM analysis, here we show that Yta7 assembles a three-tiered hexamer with a top BRD tier, a middle AAA1 tier, and a bottom AAA2 tier. Unexpectedly, the Yta7 BRD stabilizes a four-stranded β-helix, termed BRD-interacting motif (BIM), of the largely disordered N-terminal region. The BIM motif is unique to the baker's yeast, and we show both BRD and BIM contribute to nucleosome recognition. We found that Yta7 binds both acetylated and nonacetylated H3 peptides but with a higher affinity for the unmodified peptide. This property is consistent with the absence of key residues of canonical BRDs involved in acetylated peptide recognition and the role of Yta7 in general nucleosome remodeling. Interestingly, the BRD tier exists in a spiral and a flat-ring form on top of the Yta7 AAA+ hexamer. The spiral is likely in a nucleosome-searching mode because the bottom BRD blocks the entry to the AAA+ chamber. The flat ring may be in a nucleosome disassembly state because the entry is unblocked and the H3 peptide has entered the AAA+ chamber and is stabilized by the AAA1 pore loops 1 and 2. Indeed, we show that the BRD tier is a flat ring when bound to the nucleosome. Overall, our study sheds light on the nucleosome disassembly by Yta7.
Details: Solution was made fresh and detergent was added to solve a preference orientation issue.
Grid
Model: Quantifoil R2/1 / Support film - Material: GOLD / Support film - topology: CONTINUOUS
Vitrification
Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 281 K / Instrument: FEI VITROBOT MARK IV / Details: Blot 3 seconds, blot force 3.
Details
The sample was a novel chromatin remodeler and an AAA+ ATPase.
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Temperature
Min: 193.0 K / Max: 193.0 K
Image recording
Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 65.0 e/Å2 Details: A total of 75 frames was recorded for each micrograph stack.
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information
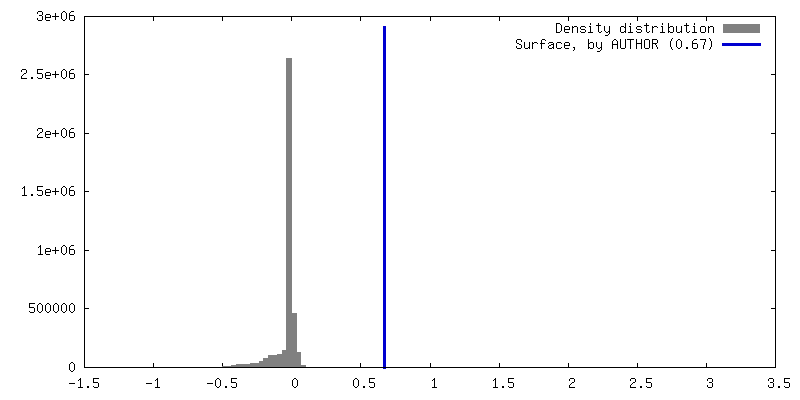
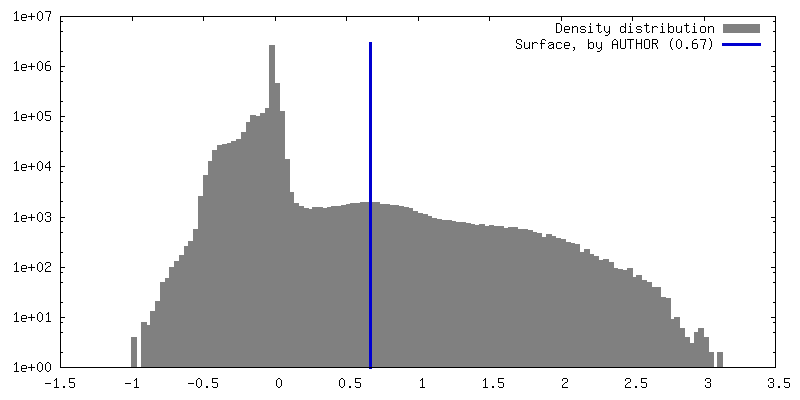
 Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_28815.png
emd_28815.png http://ftp.pdbj.org/pub/emdb/structures/EMD-28815
http://ftp.pdbj.org/pub/emdb/structures/EMD-28815









 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)
















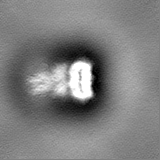
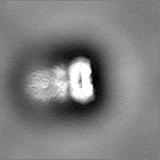


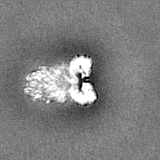
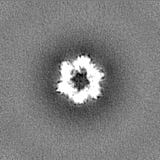


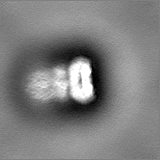

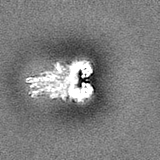
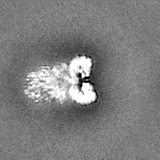



 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN