 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-2852 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
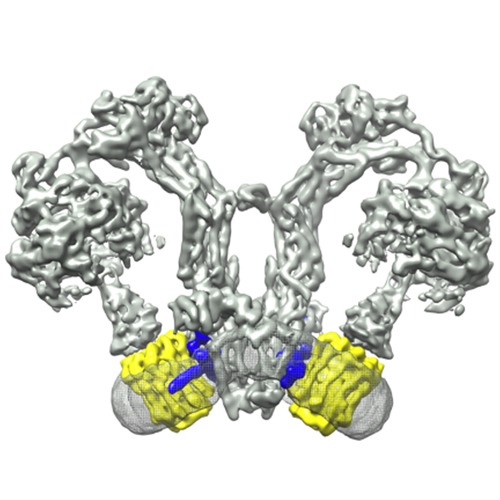
| Title | Electron cryo-microscopy of mitochondrial ATP synthase dimers | |||||||||
 Map data Map data | Reconstruction of an ATP-synthase dimer | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | ATP-synthase / mitochondria / dimers / Polytomella | |||||||||
| Biological species |  Polytomella (plant) Polytomella (plant) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 7.0 Å | |||||||||
 Authors Authors | Allegretti M / Klusch N / Mills DJ / Vonck J / Kuehlbrandt W / Davies KM | |||||||||
 Citation Citation | Journal: Nature / Year: 2015 Title: Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Authors: Matteo Allegretti / Niklas Klusch / Deryck J Mills / Janet Vonck / Werner Kühlbrandt / Karen M Davies /  Abstract: ATP, the universal energy currency of cells, is produced by F-type ATP synthases, which are ancient, membrane-bound nanomachines. F-type ATP synthases use the energy of a transmembrane ...ATP, the universal energy currency of cells, is produced by F-type ATP synthases, which are ancient, membrane-bound nanomachines. F-type ATP synthases use the energy of a transmembrane electrochemical gradient to generate ATP by rotary catalysis. Protons moving across the membrane drive a rotor ring composed of 8-15 c-subunits. A central stalk transmits the rotation of the c-ring to the catalytic F1 head, where a series of conformational changes results in ATP synthesis. A key unresolved question in this fundamental process is how protons pass through the membrane to drive ATP production. Mitochondrial ATP synthases form V-shaped homodimers in cristae membranes. Here we report the structure of a native and active mitochondrial ATP synthase dimer, determined by single-particle electron cryomicroscopy at 6.2 Å resolution. Our structure shows four long, horizontal membrane-intrinsic α-helices in the a-subunit, arranged in two hairpins at an angle of approximately 70° relative to the c-ring helices. It has been proposed that a strictly conserved membrane-embedded arginine in the a-subunit couples proton translocation to c-ring rotation. A fit of the conserved carboxy-terminal a-subunit sequence places the conserved arginine next to a proton-binding c-subunit glutamate. The map shows a slanting solvent-accessible channel that extends from the mitochondrial matrix to the conserved arginine. Another hydrophilic cavity on the lumenal membrane surface defines a direct route for the protons to an essential histidine-glutamate pair. Our results provide unique new insights into the structure and function of rotary ATP synthases and explain how ATP production is coupled to proton translocation. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_2852.map.gz | 9.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-2852-v30.xmlemd-2852.xml | 9 KB 9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_2852_fsc.xml | 9.1 KB | Display | FSC data file |
| Images | 500_500_EMD-2852.tif | 606 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2852ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2852 http://ftp.pdbj.org/pub/emdb/structures/EMD-2852ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2852 | HTTPS FTP |
-Related structure data
| Similar structure data | |
|---|---|
| EM raw data | EMPIAR-10023 (Title: Electron cryo-microscopy of ATP synthase dimers from Polytomella sp. Data size: 4.1 TB Data #1: Multi-frame micrographs (1-24 frames) aligned by motion correction (Li et al 2013) [micrographs - multiframe]) |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_2852.map.gz / Format: CCP4 / Size: 62.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of an ATP-synthase dimer | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.77 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Polytomella ATP-synthase
| Entire | Name: Polytomella ATP-synthase |
|---|---|
| Components |
|
-Supramolecule #1000: Polytomella ATP-synthase
| Supramolecule | Name: Polytomella ATP-synthase / type: sample / ID: 1000 / Oligomeric state: dimer / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 1.6 MDa |
-Macromolecule #1: Mitochondrial F-type ATP-synthase
| Macromolecule | Name: Mitochondrial F-type ATP-synthase / type: protein_or_peptide / ID: 1 / Oligomeric state: Dimer / Recombinant expression: No |
|---|---|
| Source (natural) | Organism: Polytomella (plant) / Organelle: Mitochondrion / Location in cell: Cristae membrane |
| Molecular weight | Theoretical: 1.6 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 8 Details: 50nM TRIS/HCl pH 8.0, 1mM MgCl2, 20mM NaCl, DDM 0.05% |
| Grid | Details: Quantifoil gold grid with thin carbon support, glow discharged for 2 minutes |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 75 % / Chamber temperature: 113 K / Instrument: FEI VITROBOT MARK I / Method: Blot for 8-10 seconds before plunging |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Temperature | Average: 78 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at 155,000 magnification |
| Date | Jan 30, 2014 |
| Image recording | Category: CCD / Film or detector model: FEI FALCON II (4k x 4k) / Number real images: 2829 / Average electron dose: 80 e/Å2 Details: Every image is a stack aligned by the motion correction software (Li et al., 2013) |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 104430 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.2 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 1.5 µm / Nominal magnification: 59000 |
| Sample stage | Specimen holder: Nitrogen cooled / Specimen holder model: OTHER |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
-Image processing
| Details | The particles were selected manually using EMAN boxer |
|---|---|
| CTF correction | Details: CTFFIND3 |
| Final reconstruction | Applied symmetry - Point group: C2 (2 fold cyclic) / Resolution.type: BY AUTHOR / Resolution: 7.0 Å / Resolution method: OTHER / Software - Name: EMAN, RELION, IMOD, IMAGIC / Number images used: 49600 |
| FSC plot (resolution estimation) | 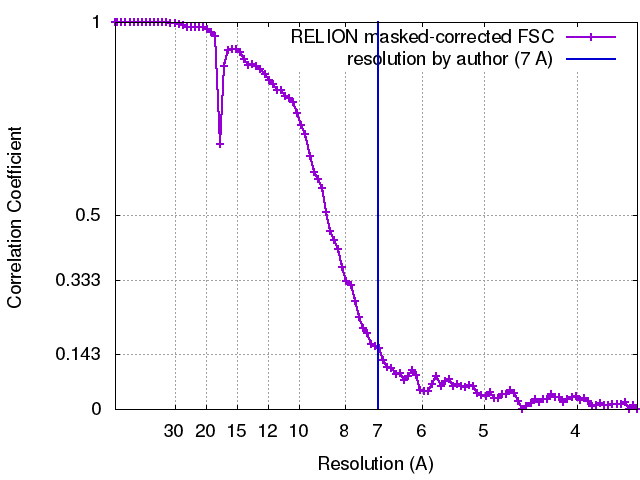 |
