 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Negative stain EM map of an MTA-HDAC-RBBP4 complex | |||||||||||||||||||||
 マップデータ マップデータ | Main map of MHR after homogeneous refinement. | |||||||||||||||||||||
 試料 試料 |
| |||||||||||||||||||||
 キーワード キーワード | Complex / Subcomplex / Deacetylase / Remodeller / Protein Binding / DNA BINDING PROTEIN | |||||||||||||||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | |||||||||||||||||||||
| 手法 | 単粒子再構成法 / ネガティブ染色法 / 解像度: 26.0 Å | |||||||||||||||||||||
 データ登録者 データ登録者 | Jackman MJ / Landsberg MJ / Viswanath S / Arvindekar S / Mackay JP | |||||||||||||||||||||
| 資金援助 |  オーストラリア, オーストラリア,  インド, 6件 インド, 6件
| |||||||||||||||||||||
 引用 引用 | ジャーナル: Protein Sci / 年: 2022 タイトル: Molecular architecture of nucleosome remodeling and deacetylase sub-complexes by integrative structure determination. 著者: Shreyas Arvindekar / Matthew J Jackman / Jason K K Low / Michael J Landsberg / Joel P Mackay / Shruthi Viswanath / 要旨: The nucleosome remodeling and deacetylase (NuRD) complex is a chromatin-modifying assembly that regulates gene expression and DNA damage repair. Despite its importance, limited structural information ...The nucleosome remodeling and deacetylase (NuRD) complex is a chromatin-modifying assembly that regulates gene expression and DNA damage repair. Despite its importance, limited structural information describing the complete NuRD complex is available and a detailed understanding of its mechanism is therefore lacking. Drawing on information from SEC-MALLS, DIA-MS, XLMS, negative-stain EM, X-ray crystallography, NMR spectroscopy, secondary structure predictions, and homology models, we applied Bayesian integrative structure determination to investigate the molecular architecture of three NuRD sub-complexes: MTA1-HDAC1-RBBP4, MTA1 -HDAC1-MBD3 , and MTA1-HDAC1-RBBP4-MBD3-GATAD2A [nucleosome deacetylase (NuDe)]. The integrative structures were corroborated by examining independent crosslinks, cryo-EM maps, biochemical assays, known cancer-associated mutations, and structure predictions from AlphaFold. The robustness of the models was assessed by jack-knifing. Localization of the full-length MBD3, which connects the deacetylase and chromatin remodeling modules in NuRD, has not previously been possible; our models indicate two different locations for MBD3, suggesting a mechanism by which MBD3 in the presence of GATAD2A asymmetrically bridges the two modules in NuRD. Further, our models uncovered three previously unrecognized subunit interfaces in NuDe: HDAC1 -MTA1 , MTA1 -MBD3 , and HDAC1 -MBD3 . Our approach also allowed us to localize regions of unknown structure, such as HDAC1 and MBD3 , thereby resulting in the most complete and robustly cross-validated structural characterization of these NuRD sub-complexes so far. | |||||||||||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_27557.map.gz | 29.7 MB |  EMDBマップデータ形式 EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-27557-v30.xmlemd-27557.xml | 21.8 KB 21.8 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_27557_fsc.xml | 11.5 KB | 表示 | FSCデータファイル |
| 画像 |  emd_27557.png emd_27557.png | 19.4 KB | ||
| マスクデータ | emd_27557_msk_1.map | 59.6 MB | マスクマップ | |
| Filedesc metadata | emd-27557.cif.gz | 5.6 KB | ||
| その他 | emd_27557_additional_1.map.gzemd_27557_half_map_1.map.gzemd_27557_half_map_2.map.gz | 29 MB 55.2 MB 55.2 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-27557ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27557 http://ftp.pdbj.org/pub/emdb/structures/EMD-27557ftp://ftp.pdbj.org/pub/emdb/structures/EMD-27557 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_27557_validation.pdf.gz | 645.7 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_27557_full_validation.pdf.gz | 645.3 KB | 表示 | |
| XML形式データ | emd_27557_validation.xml.gz | 14.7 KB | 表示 | |
| CIF形式データ | emd_27557_validation.cif.gz | 20.8 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27557ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-27557 | HTTPS FTP |
-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_27557.map.gz / 形式: CCP4 / 大きさ: 59.6 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Main map of MHR after homogeneous refinement. | ||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 2.79 Å | ||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
-マスク #1
| ファイル | emd_27557_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 投影像・断面図 |
| ||||||||||||
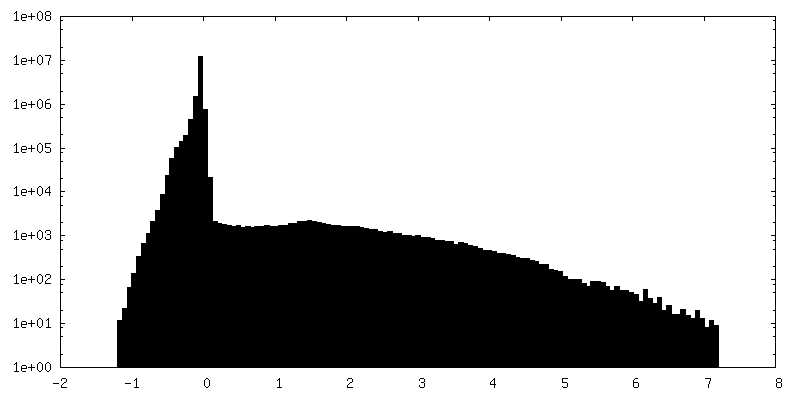
| 密度ヒストグラム |






-追加マップ: Input used in the IMP for the generation...
| ファイル | emd_27557_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Input used in the IMP for the generation of the predicted MHR model. It was derived from an independent processing run using the same dataset as the model used to generate the final map. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |





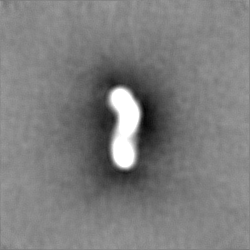
-ハーフマップ: Half map B from homogeneous refinement that produced...
| ファイル | emd_27557_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
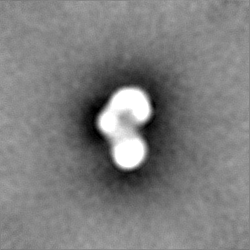
| 注釈 | Half map B from homogeneous refinement that produced final map of MHR. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |

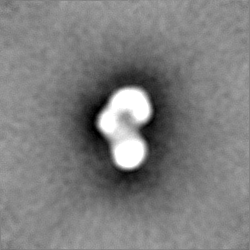


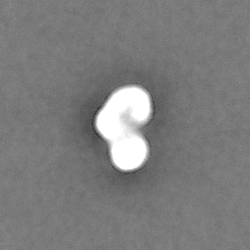
-ハーフマップ: Half map A from homogeneous refinement that produced...
| ファイル | emd_27557_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
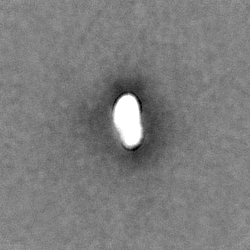
| 注釈 | Half map A from homogeneous refinement that produced final map of MHR. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |




- 試料の構成要素
試料の構成要素
-全体 : Low resolution map of the MTA1:HDAC1:RBBP7 complex from HEK Expi2...
| 全体 | 名称: Low resolution map of the MTA1:HDAC1:RBBP7 complex from HEK Expi293F cells |
|---|---|
| 要素 |
|
-超分子 #1: Low resolution map of the MTA1:HDAC1:RBBP7 complex from HEK Expi2...
| 超分子 | 名称: Low resolution map of the MTA1:HDAC1:RBBP7 complex from HEK Expi293F cells タイプ: complex / ID: 1 / 親要素: 0 詳細: Subcomplex that was generated via combination of recombinant proteins pulled down via FLAG tags from HEK Expi293F cells |
|---|---|
| 由来(天然) | 生物種: Homo sapiens (ヒト) |
-実験情報
-構造解析
| 手法 | ネガティブ染色法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.015 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 緩衝液 | pH: 7.5 構成要素:
詳細: This is the exchange buffer used for both concentrating the sample and also removing sucrose after GraFix. | ||||||||||||
| 染色 | タイプ: NEGATIVE / 材質: Uranyl Acetate 詳細: 5 uL of sample was applied to the grid and incubated for 2 minutes. The grid was then blotted and washed with 10 drops of distilled water, with liquid blotted off between each drop. The grid ...詳細: 5 uL of sample was applied to the grid and incubated for 2 minutes. The grid was then blotted and washed with 10 drops of distilled water, with liquid blotted off between each drop. The grid was then pre-stained with a drop 1% Uranyl Acetate, blotted, then stained with a drop of 1% Uranyl Acetate for 30 seconds. It was then blotted again and left to air dry at room temperature. | ||||||||||||
| グリッド | モデル: Quantifoil / 材質: COPPER / メッシュ: 400 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: CONTINUOUS / 支持フィルム - Film thickness: 5 / 前処理 - タイプ: GLOW DISCHARGE | ||||||||||||
| 詳細 | The sample was in high abundance, and appears to be well dispersed. Some aggregation is clear, and the particles upon closer inspection (such as 2D classification) are heterogeneous. This is likely due to the flexibility of the complex. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI 12 |
|---|---|
| 撮影 | フィルム・検出器のモデル: OTHER / デジタル化 - サイズ - 横: 4096 pixel / デジタル化 - サイズ - 縦: 4096 pixel / 実像数: 469 / 平均電子線量: 100.0 e/Å2 詳細: Images recorded on a DE LC1100 lens coupled CCD detector. Estimated dose. |
| 電子線 | 加速電圧: 120 kV / 電子線源: LAB6 |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / 最大 デフォーカス(公称値): 2.5 µm / 最小 デフォーカス(公称値): 1.0 µm / 倍率(公称値): 52000 |

