Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-26816: Kinetically trapped misfolded state of the Tetrahymena ribozyme -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Kinetically trapped misfolded state of the Tetrahymena ribozyme | |||||||||
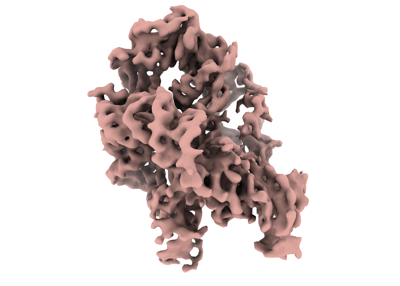
 Map data Map data | Map of misfolded state (M state) of Tetrahymena ribozyme sharpened with a B factor of 167. Refinement was performed in cryoSPARC using a 'non-uniform refinement' job. | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | ribozyme / catalytic RNA / folding intermediate / misfolded / kinetic trap / M state / Tetrahymena / RNA | |||||||||
| Biological species |   Tetrahymena thermophila (eukaryote) Tetrahymena thermophila (eukaryote) | |||||||||
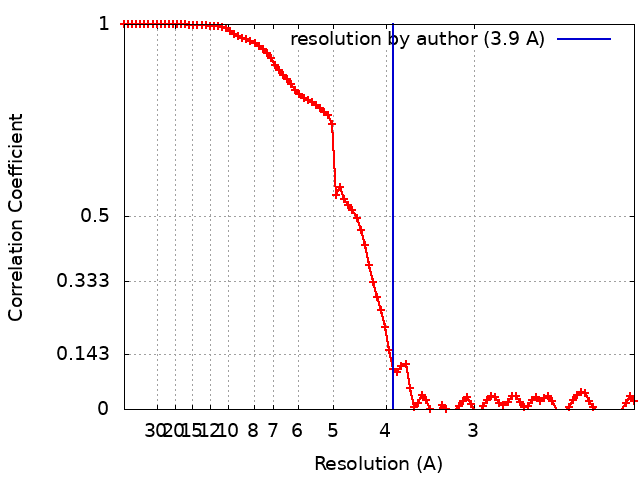
| Method | single particle reconstruction / cryo EM / Resolution: 3.9 Å | |||||||||
 Authors Authors | Bonilla SL / Vicens Q / Kieft JS | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Sci Adv / Year: 2022 Title: Cryo-EM reveals an entangled kinetic trap in the folding of a catalytic RNA. Authors: Steve L Bonilla / Quentin Vicens / Jeffrey S Kieft / Abstract: Functional RNAs fold through complex pathways that can contain misfolded "kinetic traps." A complete model of RNA folding requires understanding the formation of these misfolded states, but they are ...Functional RNAs fold through complex pathways that can contain misfolded "kinetic traps." A complete model of RNA folding requires understanding the formation of these misfolded states, but they are difficult to characterize because of their transient and potentially conformationally dynamic nature. We used cryo-electron microscopy (cryo-EM) to visualize a long-lived misfolded state in the folding pathway of the group I intron, a paradigmatic RNA structure-function model system. The structure revealed how this state forms native-like secondary structure and tertiary contacts but contains two incorrectly crossed strands, consistent with a previous model. This incorrect topology mispositions a critical catalytic domain and cannot be resolved locally as extensive refolding is required. This work provides a structural framework for interpreting decades of biochemical and functional studies and demonstrates the power of cryo-EM for the exploration of RNA folding pathways. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_26816.map.gz | 59.7 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-26816-v30.xmlemd-26816.xml | 16.3 KB 16.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_26816_fsc.xml | 8.9 KB | Display | FSC data file |
| Images | 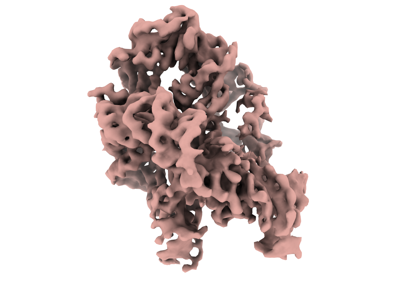 emd_26816.png emd_26816.png | 69.5 KB | ||
| Filedesc metadata | emd-26816.cif.gz | 4.6 KB | ||
| Others | emd_26816_additional_1.map.gzemd_26816_half_map_1.map.gzemd_26816_half_map_2.map.gz | 32 MB 59.3 MB 59.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-26816ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26816 http://ftp.pdbj.org/pub/emdb/structures/EMD-26816ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26816 | HTTPS FTP |
-Related structure data

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_26816.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
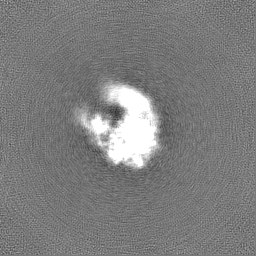
| Annotation | Map of misfolded state (M state) of Tetrahymena ribozyme sharpened with a B factor of 167. Refinement was performed in cryoSPARC using a 'non-uniform refinement' job. | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.01156 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Additional map: Unsharpened map of misfolded state (M state) of...
| File | emd_26816_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
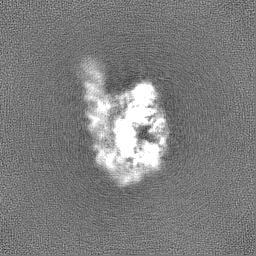
| Annotation | Unsharpened map of misfolded state (M state) of Tetrahymena ribozyme. Refinement was performed in cryoSPARC using a 'non-uniform refinement' job. | ||||||||||||
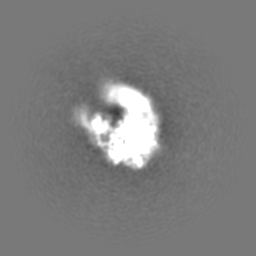
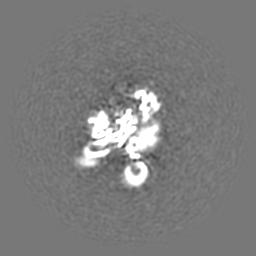
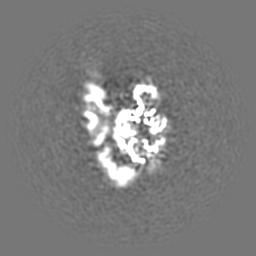
| Projections & Slices |
| ||||||||||||
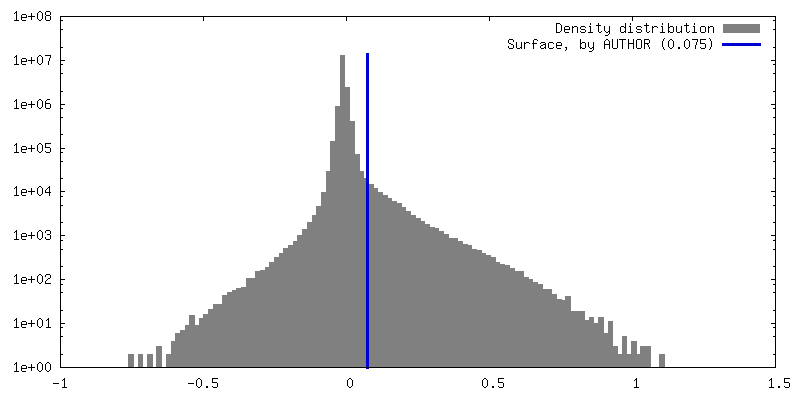
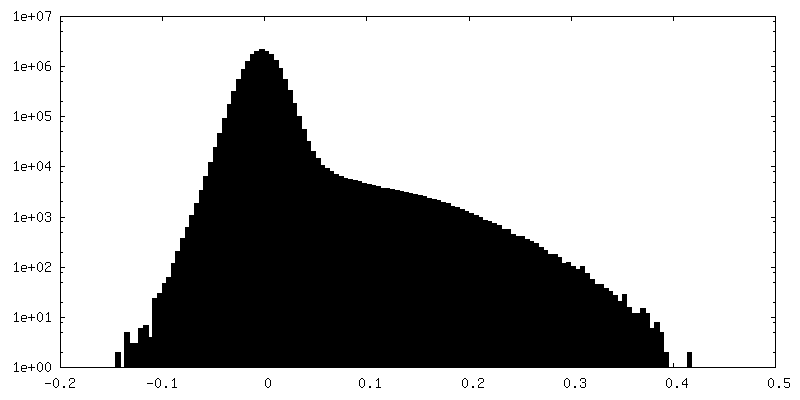
| Density Histograms |




-Half map: Half map B of misfolded state (M state)...
| File | emd_26816_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
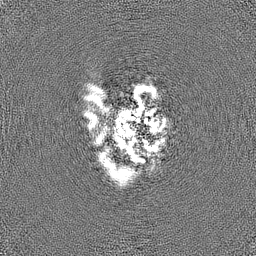
| Annotation | Half map B of misfolded state (M state) of Tetrahymena ribozyme sharpened with a B factor of 167. Refinement was performed in cryoSPARC using a 'non-uniform refinement' job. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





-Half map: Half map A of misfolded state (M state)...
| File | emd_26816_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map A of misfolded state (M state) of Tetrahymena ribozyme sharpened with a B factor of 167. Refinement was performed in cryoSPARC using a 'non-uniform refinement' job. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : apo L-21 ScaI Tetrahymena Ribozyme RNA
| Entire | Name: apo L-21 ScaI Tetrahymena Ribozyme RNA |
|---|---|
| Components |
|
-Supramolecule #1: apo L-21 ScaI Tetrahymena Ribozyme RNA
| Supramolecule | Name: apo L-21 ScaI Tetrahymena Ribozyme RNA / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all / Details: Misfolded state. |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
-Macromolecule #1: RNA (386-MER)
| Macromolecule | Name: RNA (386-MER) / type: rna / ID: 1 / Number of copies: 1 |
|---|---|
| Source (natural) | Organism: Tetrahymena thermophila (eukaryote) |
| Molecular weight | Theoretical: 125.096773 KDa |
| Sequence | String: GGAGGGAAAA GUUAUCAGGC AUGCACCUGG UAGCUAGUCU UUAAACCAAU AGAUUGCAUC GGUUUAAAAG GCAAGACCGU CAAAUUGCG GGAAAGGGGU CAACAGCCGU UCAGUACCAA GUCUCAGGGG AAACUUUGAG AUGGCCUUGC AAAGGGUAUG G UAAUAAGC ...String: GGAGGGAAAA GUUAUCAGGC AUGCACCUGG UAGCUAGUCU UUAAACCAAU AGAUUGCAUC GGUUUAAAAG GCAAGACCGU CAAAUUGCG GGAAAGGGGU CAACAGCCGU UCAGUACCAA GUCUCAGGGG AAACUUUGAG AUGGCCUUGC AAAGGGUAUG G UAAUAAGC UGACGGACAU GGUCCUAACC ACGCAGCCAA GUCCUAAGUC AACAGAUCUU CUGUUGAUAU GGAUGCAGUU CA CAGACUA AAUGUCGGUC GGGGAAGAUG UAUUCUUCUC AUAAGAUAUA GUCGGACCUC UCCUUAAUGG GAGCUAGCGG AUG AAGUGA UGCAACACUG GAGCCGCUGG GAACUAAUUU GUAUGCGAAA GUAUAUUGAU UAGUUUUGGA G GENBANK: GENBANK: X54512.1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7 Details: Buffer contained 50 mM NaMOPS, pH 7.0 and 10 mM MgCl2 |
|---|---|
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 278 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Average electron dose: 32.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.0 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |