National Institutes of Health/National Cancer Institute (NIH/NCI)
United States
Citation
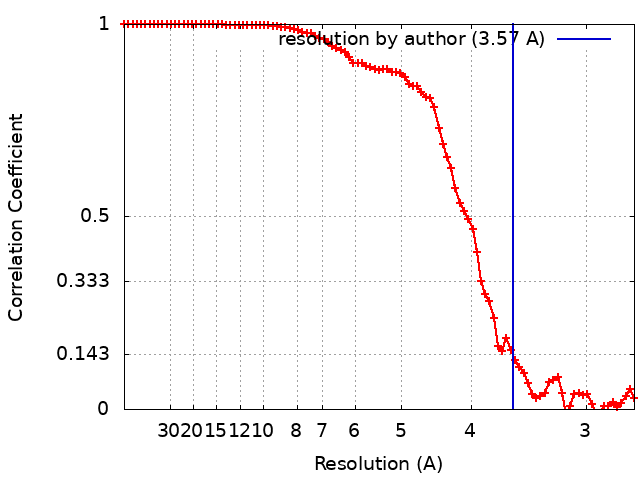
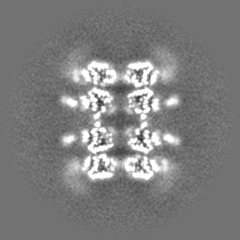
Journal: ACS Nano / Year: 2021 Title: Affinity Capture of p97 with Small-Molecule Ligand Bait Reveals a 3.6 Å Double-Hexamer Cryoelectron Microscopy Structure. Authors: Md Rejaul Hoq / Frank S Vago / Kunpeng Li / Marina Kovaliov / Robert J Nicholas / Donna M Huryn / Peter Wipf / Wen Jiang / David H Thompson / Abstract: Recent progress in the development of affinity grids for cryoelectron microscopy (cryo-EM) typically employs genetic engineering of the protein sample such as histidine or Spy tagging, immobilized ...Recent progress in the development of affinity grids for cryoelectron microscopy (cryo-EM) typically employs genetic engineering of the protein sample such as histidine or Spy tagging, immobilized antibody capture, or nonselective immobilization via electrostatic interactions or Schiff base formation. We report a powerful and flexible method for the affinity capture of target proteins for cryo-EM analysis that utilizes small-molecule ligands as bait for concentrating human target proteins directly onto the grid surface for single-particle reconstruction. This approach is demonstrated for human p97, captured using two different small-molecule high-affinity ligands of this AAA+ ATPase. Four electron density maps are revealed, each representing a p97 conformational state captured from solution, including a double-hexamer structure resolved to 3.6 Å. These results demonstrate that the noncovalent capture of protein targets on EM grids modified with high-affinity ligands can enable the structure elucidation of multiple configurational states of the target and potentially inform structure-based drug design campaigns.
History
Deposition
May 3, 2021
-
Header (metadata) release
Jun 2, 2021
-
Map release
Jun 2, 2021
-
Update
Jun 9, 2021
-
Current status
Jun 9, 2021
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Model: PELCO Ultrathin Carbon with Lacey Carbon / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 3.0 nm
Vitrification
Cryogen name: ETHANE / Chamber humidity: 80 % / Instrument: GATAN CRYOPLUNGE 3 / Details: Blot for 8 seconds before plunging..
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Image recording
Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Digitization - Dimensions - Width: 7676 pixel / Digitization - Dimensions - Height: 7420 pixel / Digitization - Sampling interval: 2.5 µm / Number real images: 1232 / Average exposure time: 10.0 sec. / Average electron dose: 43.25 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_23928.png
emd_23928.png http://ftp.pdbj.org/pub/emdb/structures/EMD-23928
http://ftp.pdbj.org/pub/emdb/structures/EMD-23928


















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)







































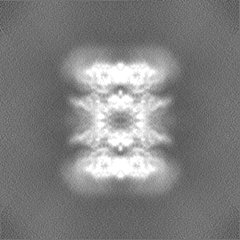

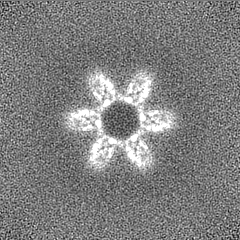
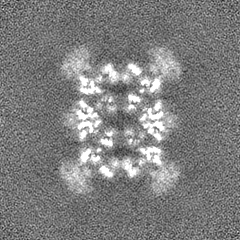

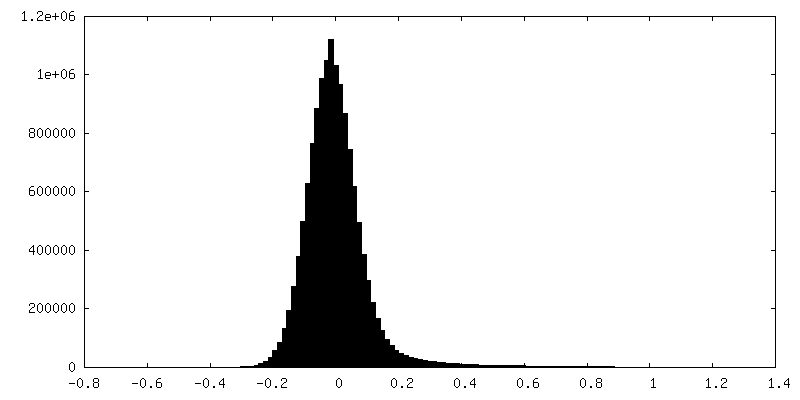
 Sample components
Sample components
 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN