 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-2268: negative stain single-particle reconstruction of conformation XXI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-2268 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | negative stain single-particle reconstruction of conformation XXI of the Ltn1 E3 ubiquitin Ligase | |||||||||
 Map data Map data | conformational snapshot XXI of Ltn1 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | conformational heterogeneity / Ltn1/Listerin / RING E3 ubiquitin ligase / translational surveillance / neurodegenerative disease | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 41.1 Å | |||||||||
 Authors Authors | Lyumkis D / Doamekpor SK / Bengtson MH / Lee JW / Toro TB / Petroski MD / Lima CD / Potter CS / Carragher B / Joazeiro CAP | |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2013 Title: Single-particle EM reveals extensive conformational variability of the Ltn1 E3 ligase. Authors: Dmitry Lyumkis / Selom K Doamekpor / Mario H Bengtson / Joong-Won Lee / Tasha B Toro / Matthew D Petroski / Christopher D Lima / Clinton S Potter / Bridget Carragher / Claudio A P Joazeiro /  Abstract: Ltn1 is a 180-kDa E3 ubiquitin ligase that associates with ribosomes and marks certain aberrant, translationally arrested nascent polypeptide chains for proteasomal degradation. In addition to its ...Ltn1 is a 180-kDa E3 ubiquitin ligase that associates with ribosomes and marks certain aberrant, translationally arrested nascent polypeptide chains for proteasomal degradation. In addition to its evolutionarily conserved large size, Ltn1 is characterized by the presence of a conserved N terminus, HEAT/ARM repeats predicted to comprise the majority of the protein, and a C-terminal catalytic RING domain, although the protein's exact structure is unknown. We used numerous single-particle EM strategies to characterize Ltn1's structure based on negative stain and vitreous ice data. Two-dimensional classifications and subsequent 3D reconstructions of electron density maps show that Ltn1 has an elongated form and presents a continuum of conformational states about two flexible hinge regions, whereas its overall architecture is reminiscent of multisubunit cullin-RING ubiquitin ligase complexes. We propose a model of Ltn1 function based on its conformational variability and flexibility that describes how these features may play a role in cotranslational protein quality control. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_2268.map.gz | 13.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-2268-v30.xmlemd-2268.xml | 13.8 KB 13.8 KB | Display Display | EMDB header |
| Images |  EMD-2268.png EMD-2268.png | 59.9 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2268ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2268 http://ftp.pdbj.org/pub/emdb/structures/EMD-2268ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2268 | HTTPS FTP |
-Validation report
| Summary document | emd_2268_validation.pdf.gz | 191.2 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_2268_full_validation.pdf.gz | 190.3 KB | Display | |
| Data in XML | emd_2268_validation.xml.gz | 5.9 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2268ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-2268 | HTTPS FTP |
-Related structure data
| Related structure data |  2248C  2249C  2250C  2251C  2252C  2253C  2254C 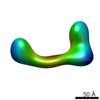 2255C  2256C  2257C  2258C  2259C  2260C  2261C  2262C  2263C  2264C  2265C  2266C  2267C  2269C C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_2268.map.gz / Format: CCP4 / Size: 15.3 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | conformational snapshot XXI of Ltn1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.18 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : snapshot XXI of Ltn1 conformer
| Entire | Name: snapshot XXI of Ltn1 conformer |
|---|---|
| Components |
|
-Supramolecule #1000: snapshot XXI of Ltn1 conformer
| Supramolecule | Name: snapshot XXI of Ltn1 conformer / type: sample / ID: 1000 / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 180 MDa / Theoretical: 180 MDa / Method: SDS-PAGE |
-Macromolecule #1: Ltn1
| Macromolecule | Name: Ltn1 / type: protein_or_peptide / ID: 1 / Name.synonym: Listerin / Number of copies: 1 / Oligomeric state: monomer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 180 MDa / Theoretical: 180 MDa |
| Recombinant expression | Organism:  |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.01 mg/mL |
|---|---|
| Buffer | pH: 8 / Details: 190 mM NaCl, 20 mM Tris, 1mM BME |
| Staining | Type: NEGATIVE Details: 3 microliters of sample at a concentration of 0.01 mg/mL was applied to a C-flat grid with 2-micron-diameter holes overlaid by thin 1.5 nm continuous carbon. The specimen was stained with 2% ...Details: 3 microliters of sample at a concentration of 0.01 mg/mL was applied to a C-flat grid with 2-micron-diameter holes overlaid by thin 1.5 nm continuous carbon. The specimen was stained with 2% uranyl formate 3 times, then let air-dry. |
| Grid | Details: c-flat grids overlaid with thin (~1.5 nm) carbon |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Temperature | Average: 298 K |
| Date | Mar 23, 2011 |
| Image recording | Category: CCD / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Number real images: 489 / Average electron dose: 15 e/Å2 |
| Tilt angle max | 0 |
| Electron beam | Acceleration voltage: 120 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 50000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 2.6 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder: single-tilt room temperature / Specimen holder model: SIDE ENTRY, EUCENTRIC / Tilt angle min: -55 |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| Details | random-conical tilt reconstruction |
|---|---|
| CTF correction | Details: each micrograph |
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 41.1 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: Spider, Appion / Number images used: 757 |
| Final two d classification | Number classes: 1 |
