Journal: J Biol Chem / Year: 2024 Title: The membrane-cytoplasmic linker defines activity of FtsH proteases in Pseudomonas aeruginosa clone C. Authors: Gina D Mawla / Shady M Kamal / Lian-Ying Cao / Pasi Purhonen / Hans Hebert / Robert T Sauer / Tania A Baker / Ute Römling / Abstract: Pandemic Pseudomonas aeruginosa clone C strains encode two inner-membrane associated ATP-dependent FtsH proteases. PaftsH1 is located on the core genome and supports cell growth and intrinsic ...Pandemic Pseudomonas aeruginosa clone C strains encode two inner-membrane associated ATP-dependent FtsH proteases. PaftsH1 is located on the core genome and supports cell growth and intrinsic antibiotic resistance, whereas PaftsH2, a xenolog acquired through horizontal gene transfer from a distantly related species, is unable to functionally replace PaftsH1. We show that purified PaFtsH2 degrades fewer substrates than PaFtsH1. Replacing the 31-amino acid-extended linker region of PaFtsH2 spanning from the C-terminal end of the transmembrane helix-2 to the first seven highly divergent residues of the cytosolic AAA+ ATPase module with the corresponding region of PaFtsH1 improves hybrid-enzyme substrate processing in vitro and enables PaFtsH2 to substitute for PaFtsH1 in vivo. Electron microscopy indicates that the identity of this linker sequence influences FtsH flexibility. We find membrane-cytoplasmic (MC) linker regions of PaFtsH1 characteristically glycine-rich compared to those from FtsH2. Consequently, introducing three glycines into the membrane-proximal end of PaFtsH2's MC linker is sufficient to elevate its activity in vitro and in vivo. Our findings establish that the efficiency of substrate processing by the two PaFtsH isoforms depends on MC linker identity and suggest that greater linker flexibility and/or length allows FtsH to degrade a wider spectrum of substrates. As PaFtsH2 homologs occur across bacterial phyla, we hypothesize that FtsH2 is a latent enzyme but may recognize specific substrates or is activated in specific contexts or biological niches. The identity of such linkers might thus play a more determinative role in the functionality of and physiological impact by FtsH proteases than previously thought.
Organelle or cellular component: Construct PFtsH2-H1-link32
-
Supramolecule #1: Construct PFtsH2-H1-link32
Supramolecule
Name: Construct PFtsH2-H1-link32 / type: organelle_or_cellular_component / ID: 1 / Parent: 0 Details: A construct containing 32-residue composed of the membrane-cytoplasmic linker (25 amino acids long) and first seven resides of the AAA+ ATPase module from PaFtsH1 with all other enzyme ...Details: A construct containing 32-residue composed of the membrane-cytoplasmic linker (25 amino acids long) and first seven resides of the AAA+ ATPase module from PaFtsH1 with all other enzyme sequences derived from PaFtsH2
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information
 Map data
Map data Sample
Sample Keywords
Keywords Pseudomonas aeruginosa SG17M (bacteria)
Pseudomonas aeruginosa SG17M (bacteria) Authors
Authors Sweden, 1 items
Sweden, 1 items  Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
Downloads & links EMDB map data format
EMDB map data format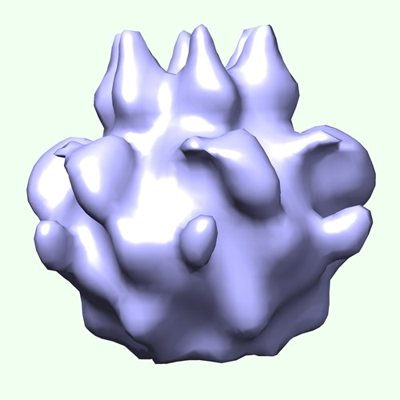 emd_18389.png
emd_18389.png http://ftp.pdbj.org/pub/emdb/structures/EMD-18389
http://ftp.pdbj.org/pub/emdb/structures/EMD-18389

 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




































 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
