 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-14591: CryoEM structure of SARS-CoV-2 spike monomer in complex with neut... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM structure of SARS-CoV-2 spike monomer in complex with neutralising antibody P008_60 | ||||||||||||||||||
 Map data Map data | DeepEMenhancer map | ||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | covid-19 / SARS-CoV-2 / spike / neutralizing antibody / immunity / VIRAL PROTEIN | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated disruption of host tissue / Maturation of spike protein / Translation of Structural Proteins / Virion Assembly and Release / host cell surface / host extracellular region / symbiont-mediated-mediated suppression of host tetherin activity / Induction of Cell-Cell Fusion / structural constituent of virion / positive regulation of viral entry into host cell ...symbiont-mediated disruption of host tissue / Maturation of spike protein / Translation of Structural Proteins / Virion Assembly and Release / host cell surface / host extracellular region / symbiont-mediated-mediated suppression of host tetherin activity / Induction of Cell-Cell Fusion / structural constituent of virion / positive regulation of viral entry into host cell / membrane fusion / host cell endoplasmic reticulum-Golgi intermediate compartment membrane / Attachment and Entry / entry receptor-mediated virion attachment to host cell / receptor-mediated virion attachment to host cell / host cell surface receptor binding / symbiont-mediated suppression of host innate immune response / endocytosis involved in viral entry into host cell / receptor ligand activity / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / symbiont entry into host cell / virion attachment to host cell / host cell plasma membrane / SARS-CoV-2 activates/modulates innate and adaptive immune responses / virion membrane / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||||||||||||||
| Biological species |  Homo sapiens (human) / Homo sapiens (human) /   Severe acute respiratory syndrome coronavirus 2 Severe acute respiratory syndrome coronavirus 2 | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.31 Å | ||||||||||||||||||
 Authors Authors | Rosa A / Pye VE / Cronin N / Cherepanov P | ||||||||||||||||||
| Funding support |  United Kingdom, 5 items United Kingdom, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Cell Rep / Year: 2022 Title: A neutralizing epitope on the SD1 domain of SARS-CoV-2 spike targeted following infection and vaccination. Authors: Jeffrey Seow / Hataf Khan / Annachiara Rosa / Valeria Calvaresi / Carl Graham / Suzanne Pickering / Valerie E Pye / Nora B Cronin / Isabella Huettner / Michael H Malim / Argyris Politis / ...Authors: Jeffrey Seow / Hataf Khan / Annachiara Rosa / Valeria Calvaresi / Carl Graham / Suzanne Pickering / Valerie E Pye / Nora B Cronin / Isabella Huettner / Michael H Malim / Argyris Politis / Peter Cherepanov / Katie J Doores / Abstract: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike is the target for neutralizing antibodies elicited following both infection and vaccination. While extensive research has shown that ...Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike is the target for neutralizing antibodies elicited following both infection and vaccination. While extensive research has shown that the receptor binding domain (RBD) and, to a lesser extent, the N-terminal domain (NTD) are the predominant targets for neutralizing antibodies, identification of neutralizing epitopes beyond these regions is important for informing vaccine development and understanding antibody-mediated immune escape. Here, we identify a class of broadly neutralizing antibodies that bind an epitope on the spike subdomain 1 (SD1) and that have arisen from infection or vaccination. Using cryo-electron microscopy (cryo-EM) and hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS), we show that SD1-specific antibody P008_60 binds an epitope that is not accessible within the canonical prefusion states of the SARS-CoV-2 spike, suggesting a transient conformation of the viral glycoprotein that is vulnerable to neutralization. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_14591.map.gz | 134.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-14591-v30.xmlemd-14591.xml | 28.4 KB 28.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_14591_fsc.xml | 11.8 KB | Display | FSC data file |
| Images | 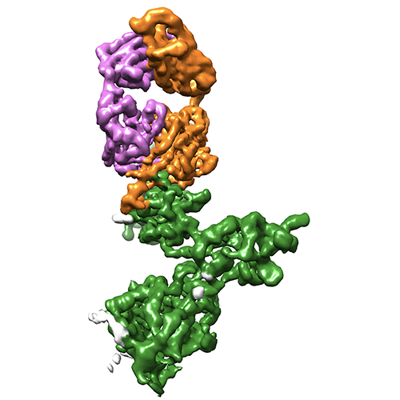 emd_14591.png emd_14591.png | 96.9 KB | ||
| Masks | emd_14591_msk_1.map | 149.9 MB | Mask map | |
| Filedesc metadata | emd-14591.cif.gz | 8.7 KB | ||
| Others | emd_14591_additional_1.map.gzemd_14591_half_map_1.map.gzemd_14591_half_map_2.map.gz | 74 MB 139.3 MB 139.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-14591ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14591 http://ftp.pdbj.org/pub/emdb/structures/EMD-14591ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14591 | HTTPS FTP |
-Related structure data
| Related structure data |  7zbuMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |




-Map
| File | Download / File: emd_14591.map.gz / Format: CCP4 / Size: 149.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | DeepEMenhancer map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.1 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_14591_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: original map, no modifications
| File | emd_14591_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | original map, no modifications | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: half map A
| File | emd_14591_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: half map B
| File | emd_14591_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Monomeric Spike glycoprotein ectodomain from SARS-CoV-2 in comple...
| Entire | Name: Monomeric Spike glycoprotein ectodomain from SARS-CoV-2 in complex with a neutralising antibody P008_60 |
|---|---|
| Components |
|
-Supramolecule #1: Monomeric Spike glycoprotein ectodomain from SARS-CoV-2 in comple...
| Supramolecule | Name: Monomeric Spike glycoprotein ectodomain from SARS-CoV-2 in complex with a neutralising antibody P008_60 type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#3 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 125 KDa |
-Macromolecule #1: Spike glycoprotein
| Macromolecule | Name: Spike glycoprotein / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Molecular weight | Theoretical: 142.269859 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: MGILPSPGMP ALLSLVSLLS VLLMGCVAET GMFVFLVLLP LVSSQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPF FSNVTWFHAI HVSGTNGTKR FDNPVLPFND GVYFASTEKS NIIRGWIFGT TLDSKTQSLL IVNNATNVVI K VCEFQFCN ...String: MGILPSPGMP ALLSLVSLLS VLLMGCVAET GMFVFLVLLP LVSSQCVNLT TRTQLPPAYT NSFTRGVYYP DKVFRSSVLH STQDLFLPF FSNVTWFHAI HVSGTNGTKR FDNPVLPFND GVYFASTEKS NIIRGWIFGT TLDSKTQSLL IVNNATNVVI K VCEFQFCN DPFLGVYYHK NNKSWMESEF RVYSSANNCT FEYVSQPFLM DLEGKQGNFK NLREFVFKNI DGYFKIYSKH TP INLVRDL PQGFSALEPL VDLPIGINIT RFQTLLALHR SYLTPGDSSS GWTAGAAAYY VGYLQPRTFL LKYNENGTIT DAV DCALDP LSETKCTLKS FTVEKGIYQT SNFRVQPTES IVRFPNITNL CPFGEVFNAT RFASVYAWNR KRISNCVADY SVLY NSASF STFKCYGVSP TKLNDLCFTN VYADSFVIRG DEVRQIAPGQ TGKIADYNYK LPDDFTGCVI AWNSNNLDSK VGGNY NYLY RLFRKSNLKP FERDISTEIY QAGSTPCNGV EGFNCYFPLQ SYGFQPTNGV GYQPYRVVVL SFELLHAPAT VCGPKK STN LVKNKCVNFN FNGLTGTGVL TESNKKFLPF QQFGRDIADT TDAVRDPQTL EILDITPCSF GGVSVITPGT NTSNQVA VL YQDVNCTEVP VAIHADQLTP TWRVYSTGSN VFQTRAGCLI GAEHVNNSYE CDIPIGAGIC ASYQTQTNSP SRASSVAS Q SIIAYTMSLG AENSVAYSNN SIAIPTNFTI SVTTEILPVS MTKTSVDCTM YICGDSTECS NLLLQYGSFC TQLNRALTG IAVEQDKNTQ EVFAQVKQIY KTPPIKDFGG FNFSQILPDP SKPSKRSFIE DLLFNKVTLA DAGFIKQYGD CLGDIAARDL ICAQKFNGL TVLPPLLTDE MIAQYTSALL AGTITSGWTF GAGAALQIPF AMQMAYRFNG IGVTQNVLYE NQKLIANQFN S AIGKIQDS LSSTASALGK LQDVVNQNAQ ALNTLVKQLS SNFGAISSVL NDILSRLDPP EAEVQIDRLI TGRLQSLQTY VT QQLIRAA EIRASANLAA TKMSECVLGQ SKRVDFCGKG YHLMSFPQSA PHGVVFLHVT YVPAQEKNFT TAPAICHDGK AHF PREGVF VSNGTHWFVT QRNFYEPQII TTDNTFVSGN CDVVIGIVNN TVYDPLQPEL DSFKEELDKY FKNHTSPDVD LGDI SGINA SVVNIQKEID RLNEVAKNLN ESLIDLQELG KYEQSGRENL YFQGGGGSGY IPEAPRDGQA YVRKDGEWVL LSTFL GHHH HHH UniProtKB: Spike glycoprotein |
-Macromolecule #2: P008_60 antibody, Heavy chain
| Macromolecule | Name: P008_60 antibody, Heavy chain / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 25.954072 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: EVQLVESGGG VVQPGRSLRL TCAASGFIFS SYGMHWVRQA PGKGLEWVAV ISYDGSYKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRAE DTAVYYCTKA DYYDFWSGYQ KTYYYYMDVW GKGTTVTISS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK D YFPEPVTV ...String: EVQLVESGGG VVQPGRSLRL TCAASGFIFS SYGMHWVRQA PGKGLEWVAV ISYDGSYKYY ADSVKGRFTI SRDNSKNTLY LQMNSLRAE DTAVYYCTKA DYYDFWSGYQ KTYYYYMDVW GKGTTVTISS ASTKGPSVFP LAPSSKSTSG GTAALGCLVK D YFPEPVTV SWNSGALTSG VHTFPAVLQS SGLYSLSSVV TVPSSSLGTQ TYICNVNHKP SNTKVDKKVG RTKHHHHHH |
-Macromolecule #3: P008_60 antibody, Light chain
| Macromolecule | Name: P008_60 antibody, Light chain / type: protein_or_peptide / ID: 3 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 23.589068 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: DIQLTQSPGT LSLSPGERAT LSCRASQSVS SSYLAWYQQK PGQAPRLLIY GTSSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYC QQYGSSPQIT FGQGTRLEIK RTVAAPSVFI FPPSDEQLKS GTASVVCLLN NFYPREAKVQ WKVDNALQSG N SQESVTEQ ...String: DIQLTQSPGT LSLSPGERAT LSCRASQSVS SSYLAWYQQK PGQAPRLLIY GTSSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYC QQYGSSPQIT FGQGTRLEIK RTVAAPSVFI FPPSDEQLKS GTASVVCLLN NFYPREAKVQ WKVDNALQSG N SQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV THQGLSSPVT KSFNRGEC |
-Macromolecule #4: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 4 / Number of copies: 8 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Macromolecule #5: 3-[5-[(4-ethyl-3-methyl-5-oxidanylidene-pyrrol-2-yl)methyl]-2-[[5...
| Macromolecule | Name: 3-[5-[(4-ethyl-3-methyl-5-oxidanylidene-pyrrol-2-yl)methyl]-2-[[5-[(3-ethyl-4-methyl-5-oxidanylidene-pyrrol-2-yl)methyl]-3-(3-hydroxy-3-oxopropyl)-4-methyl-1H-pyrrol-2-yl]methyl]-4-methyl-1H- ...Name: 3-[5-[(4-ethyl-3-methyl-5-oxidanylidene-pyrrol-2-yl)methyl]-2-[[5-[(3-ethyl-4-methyl-5-oxidanylidene-pyrrol-2-yl)methyl]-3-(3-hydroxy-3-oxopropyl)-4-methyl-1H-pyrrol-2-yl]methyl]-4-methyl-1H-pyrrol-3-yl]propanoic acid type: ligand / ID: 5 / Number of copies: 1 / Formula: 3Q9 |
|---|---|
| Molecular weight | Theoretical: 588.694 Da |
| Chemical component information |  ChemComp-3Q9: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 8 Details: 0.1% n-octyl glucoside in 150 mM NaCl, 20 mM Tris-HCl, pH8.0 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Instrument: FEI VITROBOT MARK IV / Details: blotted for 3 to 4 sec before plunging. |
| Details | SARS-CoV-2 spike ectodomain (0.5 mg/ml), supplemented 0.2 mg/ml P008_60 Fab and 0.1% n-octyl glucoside in 150 mM NaCl, 20 mM Tris-HCl, pH8.0 |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 16624 / Average exposure time: 4.1 sec. / Average electron dose: 50.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.6 µm / Nominal defocus min: 0.7000000000000001 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model |
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Details | The atomistic models of monomeric SARS-CoV-2 S1 protein and a Fab molecule, extracted from PDB entries 7A92 and 5WI9, were docked in the cryo-EM map using Chimera. The NTD and the RBD of the spike subunit were replaced with the crystal structures from PDB entries 7B62 and 7OAO, respectively. Guided by the cryo-EM map, the model was adjusted and extended interactively in Coot and refined using phenix.real_space_refine. | ||||||||
| Refinement | Space: REAL / Protocol: OTHER / Overall B value: 81 / Target criteria: Correlation coefficient | ||||||||
| Output model | PDB-7zbu: |


