 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1423: Conformational reorganization of the SARS coronavirus spike follo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1423 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Conformational reorganization of the SARS coronavirus spike following receptor binding: implications for membrane fusion. | |||||||||
 Map data Map data | SARS coronavirus spike | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  SARS coronavirus SARS coronavirus | |||||||||
| Method | single particle reconstruction / cryo EM / negative staining / Resolution: 25.5 Å | |||||||||
 Authors Authors | Beniac DR / deVarennes SL / Andonov A / He R / Booth TF | |||||||||
 Citation Citation | Journal: PLoS One / Year: 2007 Title: Conformational reorganization of the SARS coronavirus spike following receptor binding: implications for membrane fusion. Authors: Daniel R Beniac / Shauna L deVarennes / Anton Andonov / Runtao He / Tim F Booth /  Abstract: The SARS coronavirus (SARS-CoV) spike is the largest known viral spike molecule, and shares a similar function with all class 1 viral fusion proteins. Previous structural studies of membrane fusion ...The SARS coronavirus (SARS-CoV) spike is the largest known viral spike molecule, and shares a similar function with all class 1 viral fusion proteins. Previous structural studies of membrane fusion proteins have largely used crystallography of static molecular fragments, in isolation of their transmembrane domains. In this study we have produced purified, irradiated SARS-CoV virions that retain their morphology, and are fusogenic in cell culture. We used cryo-electron microscopy and image processing to investigate conformational changes that occur in the entire spike of intact virions when they bind to the viral receptor, angiotensin-converting enzyme 2 (ACE2). We have shown that ACE2 binding results in structural changes that appear to be the initial step in viral membrane fusion, and precisely localized the receptor-binding and fusion core domains within the entire spike. Furthermore, our results show that receptor binding and subsequent membrane fusion are distinct steps, and that each spike can bind up to three ACE2 molecules. The SARS-CoV spike provides an ideal model system to study receptor binding and membrane fusion in the native state, employing cryo-electron microscopy and single-particle image analysis. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1423.map.gz | 59.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1423-v30.xmlemd-1423.xml | 9.1 KB 9.1 KB | Display Display | EMDB header |
| Images | 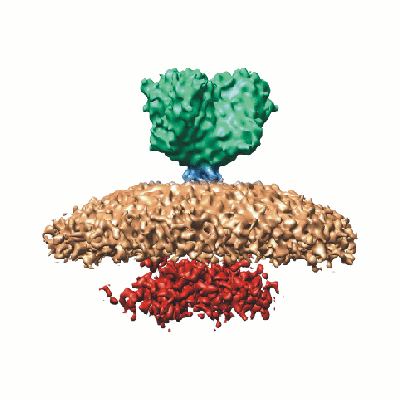 1423.gif 1423.gif | 38.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1423ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1423 http://ftp.pdbj.org/pub/emdb/structures/EMD-1423ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1423 | HTTPS FTP |
-Related structure data




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1423.map.gz / Format: CCP4 / Size: 62.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | SARS coronavirus spike | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.125 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Spike of SARS coronavirus attached to lipid envelope
| Entire | Name: Spike of SARS coronavirus attached to lipid envelope |
|---|---|
| Components |
|
-Supramolecule #1000: Spike of SARS coronavirus attached to lipid envelope
| Supramolecule | Name: Spike of SARS coronavirus attached to lipid envelope / type: sample / ID: 1000 Details: spike attached to virus was inactivated by gamma irradiation Oligomeric state: trimer - spike / Number unique components: 1 |
|---|
-Supramolecule #1: SARS coronavirus
| Supramolecule | Name: SARS coronavirus / type: virus / ID: 1 / Name.synonym: S protein / Details: spike attached to virus envelope / NCBI-ID: 227859 / Sci species name: SARS coronavirus / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: Yes / Virus empty: No / Syn species name: S protein |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) / synonym: VERTEBRATES Homo sapiens (human) / synonym: VERTEBRATES |
| Molecular weight | Experimental: 500 KDa / Theoretical: 500 KDa |
-Experimental details
-Structure determination
| Method | negative staining, cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.4 / Details: 1 times phosphate buffered saline |
|---|---|
| Staining | Type: NEGATIVE / Details: cryo, freezee plunge |
| Grid | Details: Holey carbon on 400 mesh Cu grids |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 40 % / Chamber temperature: 93 K / Instrument: REICHERT-JUNG PLUNGER / Details: Vitrification instrument: Reichert plunger / Method: blot 3 seconds before plunging |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI 20 |
|---|---|
| Temperature | Average: 93 K |
| Date | Dec 19, 2005 |
| Image recording | Category: CCD / Film or detector model: KODAK SO-163 FILM / Digitization - Sampling interval: 6.4 µm / Number real images: 63 / Average electron dose: 10 e/Å2 / Bits/pixel: 8 |
| Electron beam | Acceleration voltage: 200 kV / Electron source: LAB6 |
| Electron optics | Calibrated magnification: 29968 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 12.7 µm / Nominal defocus min: 3.3 µm / Nominal magnification: 29000 |
| Sample stage | Specimen holder: Side entry liquid nitrogen-coolde / Specimen holder model: GATAN LIQUID NITROGEN |
-Image processing
| Details | The particles were selected manually |
|---|---|
| CTF correction | Details: each particle |
| Final reconstruction | Applied symmetry - Point group: C3 (3 fold cyclic) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 25.5 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: SPIDER / Number images used: 8386 |
| Final two d classification | Number classes: 1486 |
