Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-13930: 3D reconstruction of the membrane domains of the sialic acid TRAP... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | 3D reconstruction of the membrane domains of the sialic acid TRAP transporter HiSiaQM from Haemophilus influenzae in lipid nanodiscs bound to a high affinity megabody | |||||||||
 Map data Map data | 3D reconstruction of HiSiaQM with a megabody bound to the periplasmic side | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | membrane transporter / TRAP / sialic acid / elevator / TRANSPORT PROTEIN | |||||||||
| Function / homology | TRAP transporter large membrane protein DctM / TRAP C4-dicarboxylate transport system permease DctM subunit / : / Tripartite ATP-independent periplasmic transporters, DctQ component / Tripartite ATP-independent periplasmic transporter, DctM component / transmembrane transporter activity / plasma membrane / Sialic acid TRAP transporter permease protein SiaT Function and homology information Function and homology information | |||||||||
| Biological species |  Haemophilus influenzae (bacteria) / Haemophilus influenzae (bacteria) /  | |||||||||
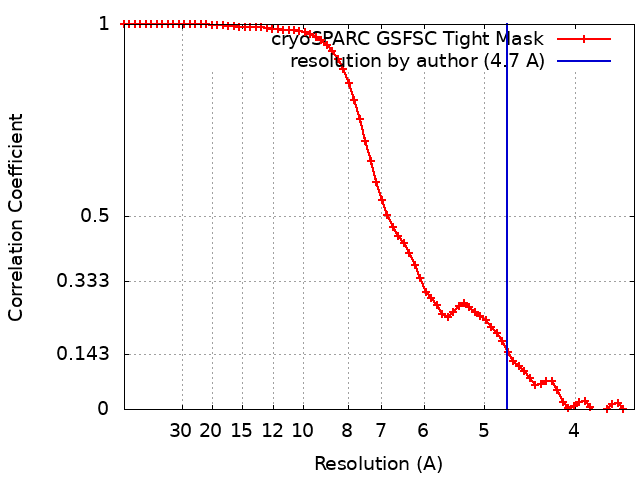
| Method | single particle reconstruction / cryo EM / Resolution: 4.7 Å | |||||||||
 Authors Authors | Peter MF / Hagelueken G | |||||||||
| Funding support |  Germany, 2 items Germany, 2 items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2022 Title: Structural and mechanistic analysis of a tripartite ATP-independent periplasmic TRAP transporter. Authors: Martin F Peter / Jan A Ruland / Peer Depping / Niels Schneberger / Emmanuele Severi / Jonas Moecking / Karl Gatterdam / Sarah Tindall / Alexandre Durand / Veronika Heinz / Jan Peter ...Authors: Martin F Peter / Jan A Ruland / Peer Depping / Niels Schneberger / Emmanuele Severi / Jonas Moecking / Karl Gatterdam / Sarah Tindall / Alexandre Durand / Veronika Heinz / Jan Peter Siebrasse / Paul-Albert Koenig / Matthias Geyer / Christine Ziegler / Ulrich Kubitscheck / Gavin H Thomas / Gregor Hagelueken /   Abstract: Tripartite ATP-independent periplasmic (TRAP) transporters are found widely in bacteria and archaea and consist of three structural domains, a soluble substrate-binding protein (P-domain), and two ...Tripartite ATP-independent periplasmic (TRAP) transporters are found widely in bacteria and archaea and consist of three structural domains, a soluble substrate-binding protein (P-domain), and two transmembrane domains (Q- and M-domains). HiSiaPQM and its homologs are TRAP transporters for sialic acid and are essential for host colonization by pathogenic bacteria. Here, we reconstitute HiSiaQM into lipid nanodiscs and use cryo-EM to reveal the structure of a TRAP transporter. It is composed of 16 transmembrane helices that are unexpectedly structurally related to multimeric elevator-type transporters. The idiosyncratic Q-domain of TRAP transporters enables the formation of a monomeric elevator architecture. A model of the tripartite PQM complex is experimentally validated and reveals the coupling of the substrate-binding protein to the transporter domains. We use single-molecule total internal reflection fluorescence (TIRF) microscopy in solid-supported lipid bilayers and surface plasmon resonance to study the formation of the tripartite complex and to investigate the impact of interface mutants. Furthermore, we characterize high-affinity single variable domains on heavy chain (VHH) antibodies that bind to the periplasmic side of HiSiaQM and inhibit sialic acid uptake, providing insight into how TRAP transporter function might be inhibited in vivo. | |||||||||
| History |
|
- Structure visualization
Structure visualization
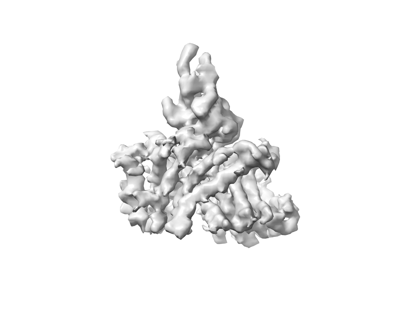
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13930.map.gz | 25.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13930-v30.xmlemd-13930.xml | 16.9 KB 16.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_13930_fsc.xml | 6.3 KB | Display | FSC data file |
| Images | 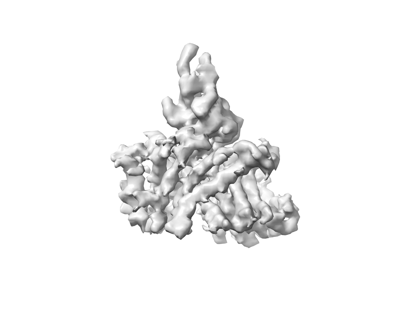 emd_13930.png emd_13930.png | 33.2 KB | ||
| Filedesc metadata | emd-13930.cif.gz | 6.2 KB | ||
| Others | emd_13930_half_map_1.map.gzemd_13930_half_map_2.map.gz | 25.1 MB 25.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13930ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13930 http://ftp.pdbj.org/pub/emdb/structures/EMD-13930ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13930 | HTTPS FTP |
-Related structure data
| Related structure data |  7qe5MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13930.map.gz / Format: CCP4 / Size: 27 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 3D reconstruction of HiSiaQM with a megabody bound to the periplasmic side | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.72 Å | ||||||||||||||||||||||||||||||||||||
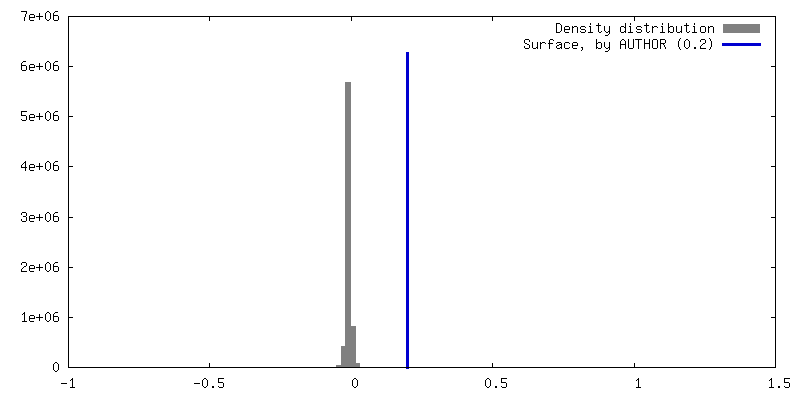
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















-Supplemental data
-Half map: Half map B
| File | emd_13930_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map B | ||||||||||||
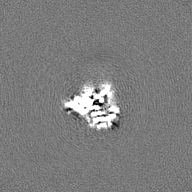
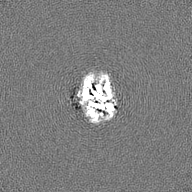
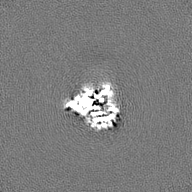
| Projections & Slices |
| ||||||||||||
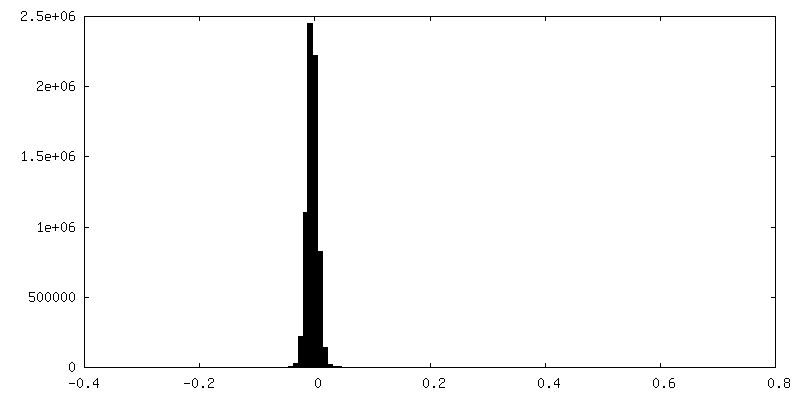
| Density Histograms |


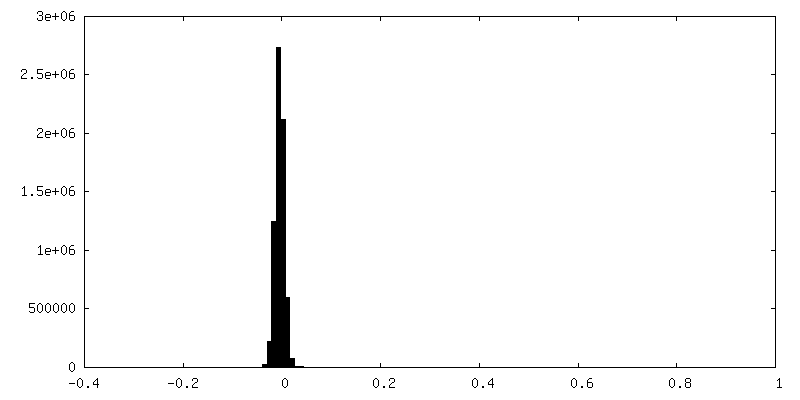
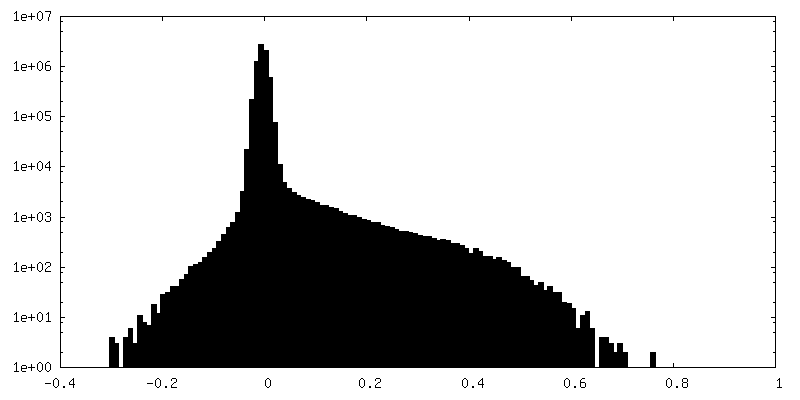
-Half map: Half map B
| File | emd_13930_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map B | ||||||||||||
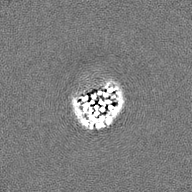
| Projections & Slices |
| ||||||||||||
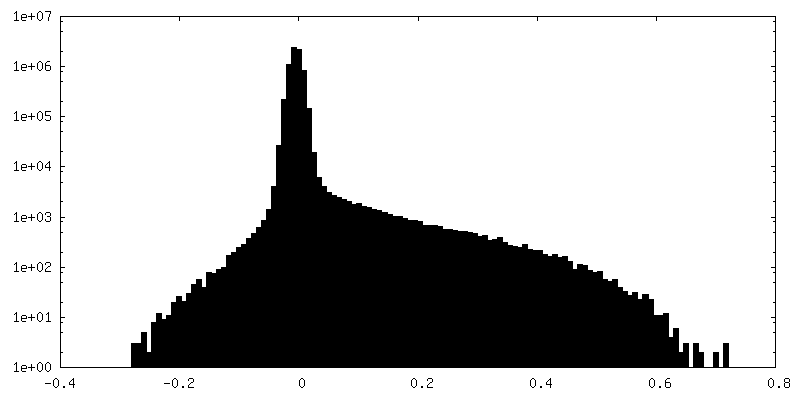
| Density Histograms |




- Sample components
Sample components
-Entire : HiSiaQM/Megabody complex
| Entire | Name: HiSiaQM/Megabody complex |
|---|---|
| Components |
|
-Supramolecule #1: HiSiaQM/Megabody complex
| Supramolecule | Name: HiSiaQM/Megabody complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Haemophilus influenzae (bacteria) |
-Macromolecule #1: Sialic acid TRAP transporter permease protein SiaT
| Macromolecule | Name: Sialic acid TRAP transporter permease protein SiaT / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Haemophilus influenzae (bacteria) / Strain: ATCC 51907 / DSM 11121 / KW20 / Rd |
| Molecular weight | Theoretical: 67.63607 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MKYINKLEEW LGGALFIAIF GILIAQILSR QVFHSPLIWS EELAKLLFVY VGMLGISVAV RKQEHVFIDF LTNLMPEKIR KFTNTFVQL LVFICIFLFI HFGIRTFNGA SFPIDALGGI SEKWIFAALP VVAILMMFRF IQAQTLNFKT GKSYLPATFF I ISAVILFA ...String: MKYINKLEEW LGGALFIAIF GILIAQILSR QVFHSPLIWS EELAKLLFVY VGMLGISVAV RKQEHVFIDF LTNLMPEKIR KFTNTFVQL LVFICIFLFI HFGIRTFNGA SFPIDALGGI SEKWIFAALP VVAILMMFRF IQAQTLNFKT GKSYLPATFF I ISAVILFA ILFFAPDWFK VLRISNYIKL GSSSVYVALL VWLIIMFIGV PVGWSLFIAT LLYFSMTRWN VVNAATEKLV YS LDSFPLL AVPFYILTGI LMNTGGITER IFNFAKALLG HYTGGMGHVN IGASLLFSGM SGSALADAGG LGQLEIKAMR DAG YDDDIC GGITAASCII GPLVPPSIAM IIYGVIANES IAKLFIAGFI PGVLITLALM AMNYRIAKKR GYPRTPKATR EQLC SSFKQ SFWAILTPLL IIGGIFSGLF SPTESAIVAA AYSVIIGKFV YKELTLKSLF NSCIEAMAIT GVVALMIMTV TFFGD MIAR EQVAMRVADV FVAVADSPLT VLIMINALLL FLGMFIDALA LQFLVLPMLI PIAMQFNIDL IFFGVMTTLN MMVGIL TPP MGMALFVVAR VGNMSVSTVT KGVLPFLIPV FVTLVLITIF PQIITFVPNL LIP UniProtKB: Sialic acid TRAP transporter permease protein SiaT |
-Macromolecule #2: Megabody3
| Macromolecule | Name: Megabody3 / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 61.534348 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MQILFQGDSH NEIPIAYGSR WIVITRGPAG HGQVQLVESG GGLVQTKTTT SVIDTTNDAQ NLLTQAQTIV NTLKDYCPIL IAKSSSSNG GTNNANTPSW QTAGGGKNSC ATFGAEFSAA SDMINNAQKI VQETQQLSAN QPKNITQPHN LNLNSPSSLT A LAQKMLKN ...String: MQILFQGDSH NEIPIAYGSR WIVITRGPAG HGQVQLVESG GGLVQTKTTT SVIDTTNDAQ NLLTQAQTIV NTLKDYCPIL IAKSSSSNG GTNNANTPSW QTAGGGKNSC ATFGAEFSAA SDMINNAQKI VQETQQLSAN QPKNITQPHN LNLNSPSSLT A LAQKMLKN AQSQAEILKL ANQVESDFNK LSSGHLKDYI GKCDASAISS ANMTMQNQKN NWGNGCAGVE ETQSLLKTSA AD FNNQTPQ INQAQNLANT LIQELGNNPF RASGGGSGGG GSGKLSDTYE QLSRLLTNDN GTNSKTSAQA INQAVNNLNE RAK TLAGGT TNSPAYQATL LALRSVLGLW NSMGYAVICG GYTKSPGENN QKDFHYTDEN GNGTTINCGG STNSNGTHSY NGTN TLKAD KNVSLSIEQY EKIHEAYQIL SKALKQAGLA PLNSKGEKLE AHVTTSYGSL RLSCTASRVT LDYHDIGWFR QAPGK EREG VSYISSSGGS TNYADSVKGR FTISRDNAKN TVYLQMNSLK PEDTAVYYCA RSSAYGSSWL NPSRYDYWGQ GTQVTV SSG GLPETGGHHH HHH |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.5 |
|---|---|
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Average electron dose: 50.213 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |