 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1390: Averaging tens to hundreds of icosahedral particle images to reso... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1390 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. | |||||||||
 Map data Map data | This is a P3A subunit of a RDV (Rice Dwarf Virus) map. It was reconstructed from the best 41 particles. For the full map, see accession code: EMD-1389. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Rice dwarf virus Rice dwarf virus | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 9.8 Å | |||||||||
 Authors Authors | Liu X / Jiang W / Jakana J / Chiu W | |||||||||
 Citation Citation | Journal: J Struct Biol / Year: 2007 Title: Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a Multi-Path Simulated Annealing optimization algorithm. Authors: Xiangan Liu / Wen Jiang / Joanita Jakana / Wah Chiu /  Abstract: Accurately determining a cryoEM particle's alignment parameters is crucial to high resolution single particle 3-D reconstruction. We developed Multi-Path Simulated Annealing, a Monte-Carlo type of ...Accurately determining a cryoEM particle's alignment parameters is crucial to high resolution single particle 3-D reconstruction. We developed Multi-Path Simulated Annealing, a Monte-Carlo type of optimization algorithm, for globally aligning the center and orientation of a particle simultaneously. A consistency criterion was developed to ensure the alignment parameters are correct and to remove some bad particles from a large pool of images of icosahedral particles. Without using any a priori model, this procedure is able to reconstruct a structure from a random initial model. Combining the procedure above with a new empirical double threshold particle selection method, we are able to pick tens of best quality particles to reconstruct a subnanometer resolution map from scratch. Using the best 62 particles of rice dwarf virus, the reconstruction reached 9.6A resolution at which four helices of the P3A subunit of RDV are resolved. Furthermore, with the 284 best particles, the reconstruction is improved to 7.9A resolution, and 21 of 22 helices and six of seven beta sheets are resolved. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera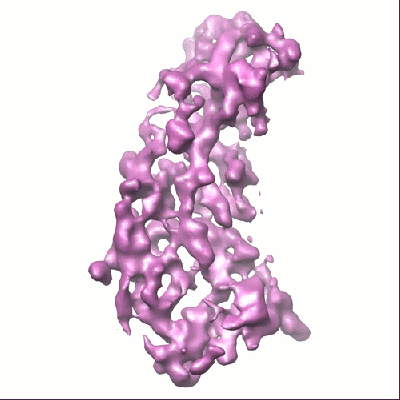
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1390.map.gz | 525.7 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1390-v30.xmlemd-1390.xml | 9 KB 9 KB | Display Display | EMDB header |
| Images |  1390.gif 1390.gif | 49.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1390ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1390 http://ftp.pdbj.org/pub/emdb/structures/EMD-1390ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1390 | HTTPS FTP |
-Related structure data
| Related structure data |  1375C  1376C  1377C  1378C  1379C  1380C  1381C  1382C  1383C  1384C  1385C  1386C  1387C  1388C  1389C C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1390.map.gz / Format: CCP4 / Size: 7.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | This is a P3A subunit of a RDV (Rice Dwarf Virus) map. It was reconstructed from the best 41 particles. For the full map, see accession code: EMD-1389. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.48906 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
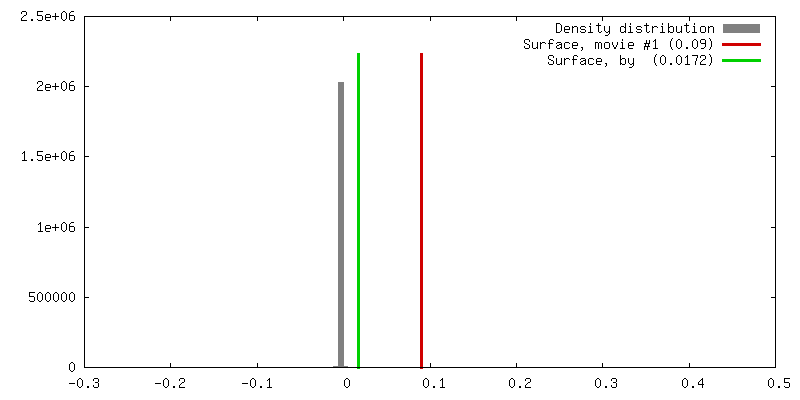
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Rice Dwarf Virus
| Entire | Name: Rice Dwarf Virus |
|---|---|
| Components |
|
-Supramolecule #1000: Rice Dwarf Virus
| Supramolecule | Name: Rice Dwarf Virus / type: sample / ID: 1000 / Number unique components: 1 |
|---|---|
| Molecular weight | Theoretical: 50 MDa |
-Supramolecule #1: Rice dwarf virus
| Supramolecule | Name: Rice dwarf virus / type: virus / ID: 1 / NCBI-ID: 10991 / Sci species name: Rice dwarf virus / Virus type: OTHER / Virus isolate: OTHER / Virus enveloped: No / Virus empty: No |
|---|---|
| Host (natural) | synonym: PLANTAE(HIGHER PLANTS) |
| Virus shell | Shell ID: 1 / Name: Inner Shell / Diameter: 550 Å / T number (triangulation number): 1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 27 % / Chamber temperature: 111 K |
|---|
- Electron microscopy
Electron microscopy
| Microscope | JEOL 4000 |
|---|---|
| Temperature | Average: 109 K |
| Details | Microscope:JEOL 4000 |
| Date | Jan 1, 1999 |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: OTHER / Digitization - Sampling interval: 4 µm / Number real images: 100 / Average electron dose: 13 e/Å2 / Bits/pixel: 32 |
| Electron beam | Acceleration voltage: 400 kV / Electron source: LAB6 |
| Electron optics | Calibrated magnification: 49495 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 4.1 mm / Nominal defocus max: 1.6 µm / Nominal defocus min: 0.3 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder: Gatan / Specimen holder model: GATAN LIQUID NITROGEN |
-Image processing
| CTF correction | Details: Each particle |
|---|---|
| Final reconstruction | Applied symmetry - Point group: I (icosahedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 9.8 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: Multi-path SA Details: The 5 alignment parameters (2 for center and 3 for orientation) were searched simultaneously for each particle image. The search is executed by a new optimizaiton algorithm, multi-path ...Details: The 5 alignment parameters (2 for center and 3 for orientation) were searched simultaneously for each particle image. The search is executed by a new optimizaiton algorithm, multi-path simulated annealing, based on Cross Common Lines. There were 4200 good particles discriminated from 4865 raw particles by a Consistency Criterion. The best 41 particles were empirically selected by a double threshold method from the 4200 good particles. The map's resolution was measured against the Known X-ray structure (PDB ID: 1UF2). Number images used: 41 |
