 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-1047 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | ATP-bound states of GroEL captured by cryo-electron microscopy. | |||||||||
 マップデータ マップデータ | GroEL(D398A)-ATP D398A has severely reduced ATPase rate (half time at least 20 min instead of 15 sec for wild type) | |||||||||
 試料 試料 |
| |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報GroEL-GroES complex / chaperonin ATPase / virion assembly / : / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat ...GroEL-GroES complex / chaperonin ATPase / virion assembly / : / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat / protein refolding / magnesium ion binding / ATP hydrolysis activity / ATP binding / identical protein binding / membrane / cytosol / cytoplasm 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 14.9 Å | |||||||||
 データ登録者 データ登録者 | Saibil HR | |||||||||
 引用 引用 | ジャーナル: Cell / 年: 2001 タイトル: ATP-bound states of GroEL captured by cryo-electron microscopy. 著者: N A Ranson / G W Farr / A M Roseman / B Gowen / W A Fenton / A L Horwich / H R Saibil /  要旨: The chaperonin GroEL drives its protein-folding cycle by cooperatively binding ATP to one of its two rings, priming that ring to become folding-active upon GroES binding, while simultaneously ...The chaperonin GroEL drives its protein-folding cycle by cooperatively binding ATP to one of its two rings, priming that ring to become folding-active upon GroES binding, while simultaneously discharging the previous folding chamber from the opposite ring. The GroEL-ATP structure, determined by cryo-EM and atomic structure fitting, shows that the intermediate domains rotate downward, switching their intersubunit salt bridge contacts from substrate binding to ATP binding domains. These observations, together with the effects of ATP binding to a GroEL-GroES-ADP complex, suggest structural models for the ATP-induced reduction in affinity for polypeptide and for cooperativity. The model for cooperativity, based on switching of intersubunit salt bridge interactions around the GroEL ring, may provide general insight into cooperativity in other ring complexes and molecular machines. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_1047.map.gz | 7.1 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-1047-v30.xmlemd-1047.xml | 10.1 KB 10.1 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  1047.gif 1047.gif | 39.2 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1047ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1047 http://ftp.pdbj.org/pub/emdb/structures/EMD-1047ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1047 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_1047_validation.pdf.gz | 247.4 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_1047_full_validation.pdf.gz | 246.6 KB | 表示 | |
| XML形式データ | emd_1047_validation.xml.gz | 5.4 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1047ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1047 | HTTPS FTP |
-関連構造データ












-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |

-マップ
| ファイル | ダウンロード / ファイル: emd_1047.map.gz / 形式: CCP4 / 大きさ: 7.8 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | GroEL(D398A)-ATP D398A has severely reduced ATPase rate (half time at least 20 min instead of 15 sec for wild type) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 1.94 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
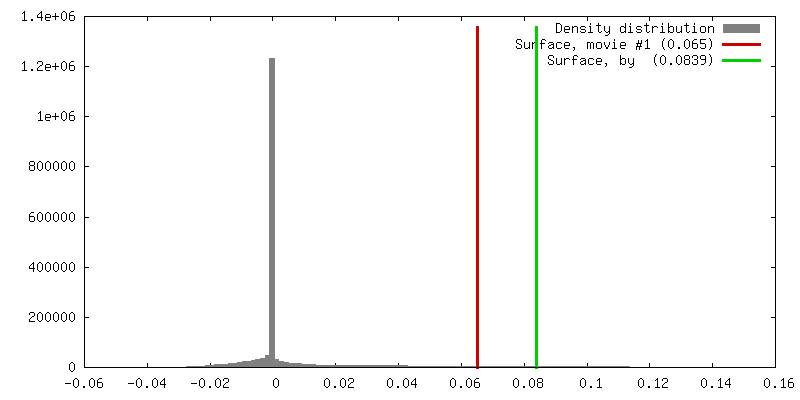
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : GroEL-ATP from E.coli
| 全体 | 名称: GroEL-ATP from E.coli |
|---|---|
| 要素 |
|
-超分子 #1000: GroEL-ATP from E.coli
| 超分子 | 名称: GroEL-ATP from E.coli / タイプ: sample / ID: 1000 / 集合状態: 14-mer / Number unique components: 1 |
|---|---|
| 分子量 | 実験値: 800 KDa / 理論値: 800 KDa |
-分子 #1: GroEL
| 分子 | 名称: GroEL / タイプ: protein_or_peptide / ID: 1 / Name.synonym: Chaperonin 詳細: D398A mutant; The Asp398Ala mutant of GroEL has severely reduced ATPase activity but can support a round of protein folding. コピー数: 14 / 集合状態: 14-mer / 組換発現: Yes |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 実験値: 800 KDa / 理論値: 800 KDa |
| 組換発現 | 生物種: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 0.8 mg/mL |
|---|---|
| 緩衝液 | pH: 7.5 / 詳細: 12.5 mM HEPES, 5 mM KCl, 5 mM MgCl2, 250 microM ATP |
| グリッド | 詳細: holey carbon film |
| 凍結 | 凍結剤: ETHANE / チャンバー内温度: 100 K / 装置: HOMEMADE PLUNGER / 詳細: Vitrification instrument: self made / 手法: Blot for 1 second before plunging |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI/PHILIPS CM200FEG/ST |
|---|---|
| 温度 | 平均: 105 K |
| 撮影 | カテゴリ: FILM / フィルム・検出器のモデル: KODAK SO-163 FILM / デジタル化 - スキャナー: ZEISS SCAI / デジタル化 - サンプリング間隔: 7 µm / 実像数: 160 / 平均電子線量: 20 e/Å2 / Od range: 1 / ビット/ピクセル: 8 |
| 電子線 | 加速電圧: 200 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 36080 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2 mm / 最大 デフォーカス(公称値): 5.0 µm / 最小 デフォーカス(公称値): 1.2 µm / 倍率(公称値): 38000 |
| 試料ステージ | 試料ホルダー: Eucentric / 試料ホルダーモデル: GATAN LIQUID NITROGEN |
-画像解析
| 詳細 | Grids were vitrified within 1 minute of mixing ATP with the GroEL mutant |
|---|---|
| CTF補正 | 詳細: CTF multiplication and merging of 2D averages |
| 最終 再構成 | 想定した対称性 - 点群: C7 (7回回転対称) / アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 14.9 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: Spider / 詳細: Filtered back projection / 使用した粒子像数: 6404 |
| 最終 角度割当 | 詳細: Full coverage around a single axis, using mainly side views |
-原子モデル構築 1
| 初期モデル | PDB ID:  1der |
|---|---|
| ソフトウェア | 名称: DockEM |
| 詳細 | Protocol: Rigid body. Manual fitting of the 3 domains of a subunit as rigid bodies using O, followed by refinement with DockEM |
| 精密化 | 空間: REAL / プロトコル: RIGID BODY FIT |
| 得られたモデル |  PDB-2c7e: |
