- EMDB-0443: Vps4 with Cyclic Peptide Bound in the Central Pore -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-0443
Title
Vps4 with Cyclic Peptide Bound in the Central Pore
Map data
Vps4 with Cyclic Peptide Bound in the Central Pore
Sample
Complex: Vps4p-Cyclic Peptide complex
Protein or peptide: Vacuolar protein sorting-associated protein 4
Protein or peptide: Designed Cyclic Peptide
Ligand: ADENOSINE-5'-DIPHOSPHATE
Ligand: BERYLLIUM TRIFLUORIDE ION
Ligand: MAGNESIUM ION
Keywords
Vps4 / ESCRT / Vta1 / AAA ATPase / TRANSPORT PROTEIN / TRANSPORT PROTEIN-PEPTIDE complex
Function / homology
Function and homology information
ESCRT IV complex / Sealing of the nuclear envelope (NE) by ESCRT-III / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / : / protein retention in Golgi apparatus / Endosomal Sorting Complex Required For Transport (ESCRT) / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly ...ESCRT IV complex / Sealing of the nuclear envelope (NE) by ESCRT-III / late endosome to lysosome transport via multivesicular body sorting pathway / intralumenal vesicle formation / : / protein retention in Golgi apparatus / Endosomal Sorting Complex Required For Transport (ESCRT) / late endosome to vacuole transport via multivesicular body sorting pathway / sterol metabolic process / nuclear membrane reassembly / multivesicular body sorting pathway / vacuole organization / midbody abscission / membrane fission / multivesicular body assembly / late endosome to vacuole transport / ubiquitin-dependent protein catabolic process via the multivesicular body sorting pathway / plasma membrane repair / reticulophagy / endosomal transport / ATPase complex / nucleus organization / autophagosome maturation / nuclear pore / macroautophagy / autophagy / protein transport / midbody / endosome / endoplasmic reticulum / protein homodimerization activity / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / plasma membrane / cytoplasm Similarity search - Function
Vacuolar protein sorting-associated protein 4, MIT domain / MIT (microtubule interacting and transport) domain / MIT domain superfamily / Vps4 oligomerisation, C-terminal / MIT domain / Microtubule Interacting and Trafficking molecule domain / : / Vps4 C terminal oligomerisation domain / AAA ATPase, AAA+ lid domain / AAA+ lid domain ...Vacuolar protein sorting-associated protein 4, MIT domain / MIT (microtubule interacting and transport) domain / MIT domain superfamily / Vps4 oligomerisation, C-terminal / MIT domain / Microtubule Interacting and Trafficking molecule domain / : / Vps4 C terminal oligomerisation domain / AAA ATPase, AAA+ lid domain / AAA+ lid domain / ATPase, AAA-type, conserved site / AAA-protein family signature. / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI)
P50 GM082545
United States
National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI)
R01 GM112080
United States
National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI)
P41 GM103310
United States
Citation
Journal: Elife / Year: 2019 Title: Structure of Vps4 with circular peptides and implications for translocation of two polypeptide chains by AAA+ ATPases. Authors: Han Han / James M Fulcher / Venkata P Dandey / Janet H Iwasa / Wesley I Sundquist / Michael S Kay / Peter S Shen / Christopher P Hill / Abstract: Many AAA+ ATPases form hexamers that unfold protein substrates by translocating them through their central pore. Multiple structures have shown how a helical assembly of subunits binds a single ...Many AAA+ ATPases form hexamers that unfold protein substrates by translocating them through their central pore. Multiple structures have shown how a helical assembly of subunits binds a single strand of substrate, and indicate that translocation results from the ATP-driven movement of subunits from one end of the helical assembly to the other end. To understand how more complex substrates are bound and translocated, we demonstrated that linear and cyclic versions of peptides bind to the AAA+ ATPase Vps4 with similar affinities, and determined cryo-EM structures of cyclic peptide complexes. The peptides bind in a hairpin conformation, with one primary strand equivalent to the single chain peptide ligands, while the second strand returns through the translocation pore without making intimate contacts with Vps4. These observations indicate a general mechanism by which AAA+ ATPases may translocate a variety of substrates that include extended chains, hairpins, and crosslinked polypeptide chains.
History
Deposition
Dec 14, 2018
-
Header (metadata) release
Apr 24, 2019
-
Map release
Aug 21, 2019
-
Update
Mar 20, 2024
-
Current status
Mar 20, 2024
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Name: MAGNESIUM ION / type: ligand / ID: 5 / Number of copies: 4 / Formula: MG
Molecular weight
Theoretical: 24.305 Da
-
Experimental details
-
Structure determination
Method
cryo EM
Processing
single particle reconstruction
Aggregation state
particle
-
Sample preparation
Buffer
pH: 7.4
Grid
Details: unspecified
Vitrification
Cryogen name: ETHANE / Instrument: SPOTITON
-
Electron microscopy
Microscope
FEI TITAN KRIOS
Image recording
Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Digitization - Frames/image: 2-50 / Average exposure time: 0.2 sec. / Average electron dose: 1.5336 e/Å2
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Electron optics
Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD
Experimental equipment
Model: Titan Krios / Image courtesy: FEI Company
-
Image processing
Startup model
Type of model: EMDB MAP
Final reconstruction
Resolution.type: BY AUTHOR / Resolution: 3.6 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION / Number images used: 237480
Initial angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: RELION
Final angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: RELION
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors United States, 3 items
United States, 3 items  Citation
Citation Structure visualization
Structure visualization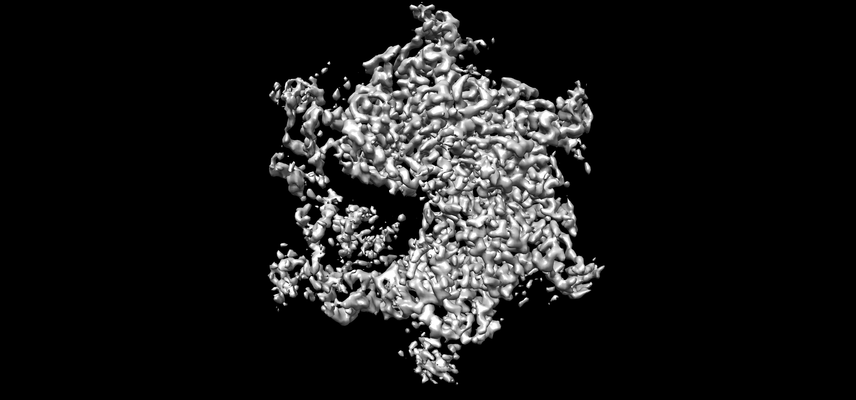
 Downloads & links
Downloads & links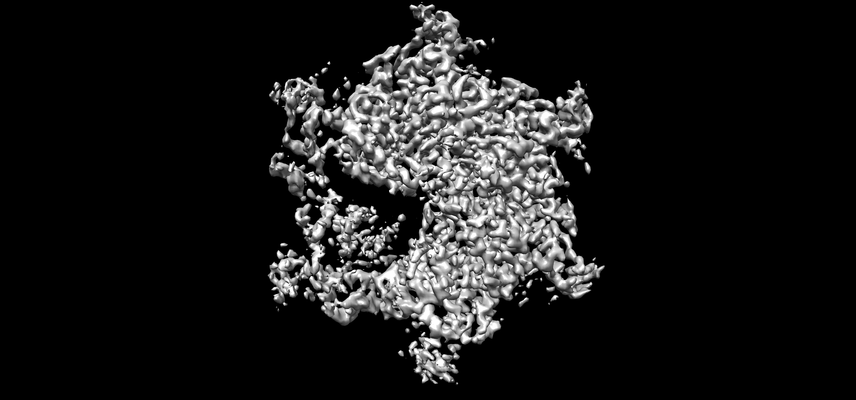 emd_0443.png
emd_0443.png http://ftp.pdbj.org/pub/emdb/structures/EMD-0443
http://ftp.pdbj.org/pub/emdb/structures/EMD-0443



















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
