 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9xko | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | High-resolution cryo-EM structure of Maltose Binding Protein | |||||||||||||||||||||||||||
 Components Components | Maltose/maltodextrin-binding periplasmic protein | |||||||||||||||||||||||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / small protein-ligand complex | |||||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationdetection of maltose stimulus / maltose transport complex / carbohydrate transport / carbohydrate transmembrane transporter activity / maltose binding / maltose transport / maltodextrin transmembrane transport / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / ATP-binding cassette (ABC) transporter complex / cell chemotaxis ...detection of maltose stimulus / maltose transport complex / carbohydrate transport / carbohydrate transmembrane transporter activity / maltose binding / maltose transport / maltodextrin transmembrane transport / ATP-binding cassette (ABC) transporter complex, substrate-binding subunit-containing / ATP-binding cassette (ABC) transporter complex / cell chemotaxis / outer membrane-bounded periplasmic space / periplasmic space / DNA damage response / membrane Similarity search - Function | |||||||||||||||||||||||||||
| Biological species |  | |||||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.35 Å | |||||||||||||||||||||||||||
 Authors Authors | Park, K. / Yoo, Y. / Jeon, H. / Choi, K. / Kwon, E. / Lim, H. / Kim, D.Y. / No, K.T. | |||||||||||||||||||||||||||
| Funding support | 1items
| |||||||||||||||||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2026 Title: High-resolution cryo-EM structures of small protein-ligand complexes near the theoretical size limit. Authors: Kunwoong Park / Youngki Yoo / Hyunbum Jeon / Kiju Choi / Hanseong Kim / Eunju Kwon / Hyun-Ho Lim / Dong Young Kim / Kyoung Tai No /  Abstract: Cryo-electron microscopy (cryo-EM) is a widely used technique for determining macromolecular structures at near-atomic resolution. The theoretical lower limit of particle sizes suitable for cryo-EM ...Cryo-electron microscopy (cryo-EM) is a widely used technique for determining macromolecular structures at near-atomic resolution. The theoretical lower limit of particle sizes suitable for cryo-EM structural analysis is estimated to be 38 kDa; typical constraints involve factors such as image contrast and particle alignment accuracy. In this study, we present cryo-EM structures of two protein-ligand complexes near this lower size threshold. First, the structure of the maltose-binding protein complexed with maltose, with a structurally ordered mass of 40.8 kDa, was determined at a resolution of 2.4 Å; both the maltose and water molecules were clearly identified in this structure. The second structure was the kinase domain of human PLK1 complexed with onvansertib, with a structurally ordered mass of 31.6 kDa, below the theoretical 38 kDa limit; this domain was determined at a resolution of 3.4 Å using a gold-supported grid in the presence of β-octyl-glucoside. The density map clearly shows the backbone of PLK1 secondary structure, and the onvansertib. These results demonstrate that cryo-EM can be effectively employed to determine structures of small proteins or domains, and to perform structure-based drug screening for small proteins, without requiring structural fiducials for particle alignment. | |||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9xko.cif.gz | 91.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9xko.ent.gz | 64.3 KB | Display | PDB format |
| PDBx/mmJSON format | 9xko.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xk/9xkoftp://data.pdbj.org/pub/pdb/validation_reports/xk/9xko | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  66973MC  22rdC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 43819.270 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: A313V substitution reflects the MBP sequence encoded in the pMAL-c5X expression vector backbone Source: (gene. exp.) |
|---|---|
| #2: Polysaccharide | alpha-D-glucopyranose-(1-4)-alpha-D-glucopyranose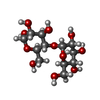  Source method: isolated from a genetically manipulated source Details: oligosaccharide / References: alpha-maltose |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: High-resolution cryo-EM structure of Maltosee Binding Protein Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 7.5 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Cryogen name: ETHANE / Humidity: 100 % |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal defocus max: 1800 nm / Nominal defocus min: 400 nm / Alignment procedure: ZEMLIN TABLEAU |
| Specimen holder | Cryogen: NITROGEN |
| Image recording | Electron dose: 60 e/Å2 / Film or detector model: FEI FALCON IV (4k x 4k) |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING ONLY | ||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 8352733 | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.35 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 509884 / Symmetry type: POINT | ||||||||||||||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT | ||||||||||||||||||||||||
| Atomic model building | PDB-ID: 1FQC Accession code: 1FQC / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||
| Refinement | Highest resolution: 2.35 Å Stereochemistry target values: REAL-SPACE (WEIGHTED MAP SUM AT ATOM CENTERS) | ||||||||||||||||||||||||
| Refine LS restraints |
|

