 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7z2g: Cryo-EM structure of HIV-1 reverse transcriptase with a DNA aptam... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7z2g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of HIV-1 reverse transcriptase with a DNA aptamer in complex with doravirine | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / Reverse transcriptase / RT-aptamer complex / non-nucleoside inhibitor / NNRTI | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / viral penetration into host nucleus / establishment of integrated proviral latency ...HIV-1 retropepsin / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / host multivesicular body / DNA integration / viral genome integration into host DNA / RNA-directed DNA polymerase / viral penetration into host nucleus / establishment of integrated proviral latency / RNA stem-loop binding / RNA-directed DNA polymerase activity / host cell / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / symbiont-mediated suppression of host gene expression / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / Hydrolases; Acting on ester bonds / aspartic-type endopeptidase activity / DNA-directed DNA polymerase activity / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding / membrane Similarity search - Function | ||||||
| Biological species |  Human immunodeficiency virus type 1 BH10 Human immunodeficiency virus type 1 BH10synthetic construct (others) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.65 Å | ||||||
 Authors Authors | Singh, A.K. / Das, K. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2022 Title: Cryo-EM structures of wild-type and E138K/M184I mutant HIV-1 RT/DNA complexed with inhibitors doravirine and rilpivirine. Authors: Abhimanyu K Singh / Brent De Wijngaert / Marc Bijnens / Kris Uyttersprot / Hoai Nguyen / Sergio E Martinez / Dominique Schols / Piet Herdewijn / Christophe Pannecouque / Eddy Arnold / Kalyan Das /   Abstract: Structures trapping a variety of functional and conformational states of HIV-1 reverse transcriptase (RT) have been determined by X-ray crystallography. These structures have played important roles ...Structures trapping a variety of functional and conformational states of HIV-1 reverse transcriptase (RT) have been determined by X-ray crystallography. These structures have played important roles in explaining the mechanisms of catalysis, inhibition, and drug resistance and in driving drug design. However, structures of several desired complexes of RT could not be obtained even after many crystallization or crystal soaking experiments. The ternary complexes of doravirine and rilpivirine with RT/DNA are such examples. Structural study of HIV-1 RT by single-particle cryo-electron microscopy (cryo-EM) has been challenging due to the enzyme's relatively smaller size and higher flexibility. We optimized a protocol for rapid structure determination of RT complexes by cryo-EM and determined six structures of wild-type and E138K/M184I mutant RT/DNA in complexes with the nonnucleoside inhibitors rilpivirine, doravirine, and nevirapine. RT/DNA/rilpivirine and RT/DNA/doravirine complexes have structural differences between them and differ from the typical conformation of nonnucleoside RT inhibitor (NNRTI)-bound RT/double-stranded DNA (dsDNA), RT/RNA-DNA, and RT/dsRNA complexes; the primer grip in RT/DNA/doravirine and the YMDD motif in RT/DNA/rilpivirine have large shifts. The DNA primer 3'-end in the doravirine-bound structure is positioned at the active site, but the complex is in a nonproductive state. In the mutant RT/DNA/rilpivirine structure, I184 is stacked with the DNA such that their relative positioning can influence rilpivirine in the pocket. Simultaneously, E138K mutation opens the NNRTI-binding pocket entrance, potentially contributing to a faster rate of rilpivirine dissociation by E138K/M184I mutant RT, as reported by an earlier kinetic study. These structural differences have implications for understanding molecular mechanisms of drug resistance and for drug design. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7z2g.cif.gz | 194.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7z2g.ent.gz | 154.6 KB | Display | PDB format |
| PDBx/mmJSON format | 7z2g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7z2g_validation.pdf.gz | 1.2 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7z2g_full_validation.pdf.gz | 1.2 MB | Display | |
| Data in XML | 7z2g_validation.xml.gz | 38.4 KB | Display | |
| Data in CIF | 7z2g_validation.cif.gz | 57.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/z2/7z2gftp://data.pdbj.org/pub/pdb/validation_reports/z2/7z2g | HTTPS FTP |
-Related structure data
| Related structure data |  14465MC  7z24C  7z29C  7z2dC  7z2eC  7z2hC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |





-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 64037.383 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: P66 subunit Source: (gene. exp.) Human immunodeficiency virus type 1 BH10Strain: isolate BH10 / Gene: gag-pol / Plasmid: pETDuet-1 / Production host:  References: UniProt: P03366, RNA-directed DNA polymerase, DNA-directed DNA polymerase, retroviral ribonuclease H, exoribonuclease H |
|---|---|
| #2: Protein | Mass: 50039.488 Da / Num. of mol.: 1 / Fragment: P51 subunit Source method: isolated from a genetically manipulated source Details: P51 subunit Source: (gene. exp.) Human immunodeficiency virus type 1 BH10Gene: gag-pol / Plasmid: pETDuet-1 / Production host: References: UniProt: P03366, RNA-directed DNA polymerase, DNA-directed DNA polymerase, retroviral ribonuclease H, exoribonuclease H |
| #3: DNA chain | Mass: 11739.513 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) |
| #4: Chemical | ChemComp-2KW / 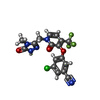  Mass: 425.749 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H11ClF3N5O3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, inhibitor*YM Mass: 425.749 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H11ClF3N5O3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, inhibitor*YM |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight |
| ||||||||||||||||||||||||
| Source (natural) |
| ||||||||||||||||||||||||
| Source (recombinant) |
| ||||||||||||||||||||||||
| Buffer solution | pH: 8 | ||||||||||||||||||||||||
| Buffer component |
| ||||||||||||||||||||||||
| Specimen | Conc.: 0.3 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | ||||||||||||||||||||||||
| Specimen support | Grid material: GOLD / Grid mesh size: 200 divisions/in. / Grid type: Quantifoil R1.2/1.3 | ||||||||||||||||||||||||
| Vitrification | Instrument: LEICA EM GP / Cryogen name: ETHANE / Humidity: 95 % / Chamber temperature: 281 K |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: TFS GLACIOS |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 150000 X / Nominal defocus max: 2200 nm / Nominal defocus min: 800 nm / Calibrated defocus min: 500 nm / Calibrated defocus max: 2500 nm / Cs: 2.7 mm / C2 aperture diameter: 50 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN |
| Image recording | Average exposure time: 55 sec. / Electron dose: 40 e/Å2 / Detector mode: COUNTING / Film or detector model: FEI FALCON III (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 1352 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: NONE | ||||||||||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 1235740 | ||||||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.65 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 178579 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||
| Atomic model building | B value: 194.3 / Protocol: FLEXIBLE FIT / Space: REAL / Target criteria: Correlation coefficient | ||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 5HP1 |

