 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7ofh: CryoEM structure of the outer membrane secretin pore pIV from the... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7ofh | ||||||
|---|---|---|---|---|---|---|---|
| Title | CryoEM structure of the outer membrane secretin pore pIV from the f1 filamentous bacteriophage. | ||||||
 Components Components | Virion export protein | ||||||
 Keywords Keywords | VIRAL PROTEIN / Secretin Outer membrane Virion export | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Enterobacteria phage f1 (virus) Enterobacteria phage f1 (virus) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.7 Å | ||||||
 Authors Authors | Conners, R. / Gold, V.A.M. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2021 Title: CryoEM structure of the outer membrane secretin channel pIV from the f1 filamentous bacteriophage. Authors: Rebecca Conners / Mathew McLaren / Urszula Łapińska / Kelly Sanders / M Rhia L Stone / Mark A T Blaskovich / Stefano Pagliara / Bertram Daum / Jasna Rakonjac / Vicki A M Gold /   Abstract: The Ff family of filamentous bacteriophages infect gram-negative bacteria, but do not cause lysis of their host cell. Instead, new virions are extruded via the phage-encoded pIV protein, which has ...The Ff family of filamentous bacteriophages infect gram-negative bacteria, but do not cause lysis of their host cell. Instead, new virions are extruded via the phage-encoded pIV protein, which has homology with bacterial secretins. Here, we determine the structure of pIV from the f1 filamentous bacteriophage at 2.7 Å resolution by cryo-electron microscopy, the first near-atomic structure of a phage secretin. Fifteen f1 pIV subunits assemble to form a gated channel in the bacterial outer membrane, with associated soluble domains projecting into the periplasm. We model channel opening and propose a mechanism for phage egress. By single-cell microfluidics experiments, we demonstrate the potential for secretins such as pIV to be used as adjuvants to increase the uptake and efficacy of antibiotics in bacteria. Finally, we compare the f1 pIV structure to its homologues to reveal similarities and differences between phage and bacterial secretins. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7ofh.cif.gz | 693.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7ofh.ent.gz | 562.7 KB | Display | PDB format |
| PDBx/mmJSON format | 7ofh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/of/7ofhftp://data.pdbj.org/pub/pdb/validation_reports/of/7ofh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  12874MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10807 (Title: CryoEM structure of the outer membrane secretin channel pIV from the f1 filamentous bacteriophage Data size: 7.0 TB Data #1: Micrographs of the secretin protein, pIV, from the f1 filamentous bacteriophage (dataset1) [micrographs - multiframe] Data #2: Micrographs of the secretin protein, pIV, from the f1 filamentous bacteriophage (dataset2) [micrographs - multiframe]) |





-Links
 PDBj
PDBj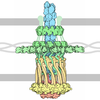
- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 44657.707 Da / Num. of mol.: 15 / Mutation: S318I Source method: isolated from a genetically manipulated source Details: S9P, D49N, I66N are naturally occurring polymorphisms. Source: (gene. exp.) Enterobacteria phage f1 (virus) / Gene: IV / Variant: S9P, D49N, I66N / Plasmid: pPMR132 / Production host:  #2: Chemical | ChemComp-CPS / 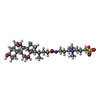  Mass: 614.877 Da / Num. of mol.: 30 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YM Mass: 614.877 Da / Num. of mol.: 30 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YMHas ligand of interest | N | Nonpolymer details | Residue 501 is full-length CHAPS, and residue 502 has been modeled as the CHAPS aromatic rings ...Residue 501 is full-length CHAPS, and residue 502 has been modeled as the CHAPS aromatic rings only, due to there being no clear density for the rest of the CHAPS molecule. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Virion export protein pIV / Type: ORGANELLE OR CELLULAR COMPONENT / Details: From Inoviridae sp. f1 / Entity ID: #1 / Source: RECOMBINANT | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.6697 MDa / Experimental value: NO | |||||||||||||||||||||||||
| Source (natural) | Organism: Inoviridae sp. (virus) | |||||||||||||||||||||||||
| Source (recombinant) | Organism: | |||||||||||||||||||||||||
| Buffer solution | pH: 8 | |||||||||||||||||||||||||
| Buffer component |
| |||||||||||||||||||||||||
| Specimen | Conc.: 0.7 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | |||||||||||||||||||||||||
| Specimen support | Grid mesh size: 300 divisions/in. / Grid type: PELCO Ultrathin Carbon with Lacey Carbon | |||||||||||||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 277 K / Details: blot force 0, blot time 4sec |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: SPOT SCAN FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: SPOT SCAN |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 81000 X |
| Image recording | Average exposure time: 3.52 sec. / Electron dose: 42.059 e/Å2 / Film or detector model: GATAN K3 (6k x 4k) |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C15 (15 fold cyclic) | ||||||||||||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.7 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 111679 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | Protocol: RIGID BODY FIT / Space: REAL | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 5W68 Accession code: 5W68 / Source name: PDB / Type: experimental model |

