 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-14842: Cryo-EM structure of the canine distemper virus tetrameric attach... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
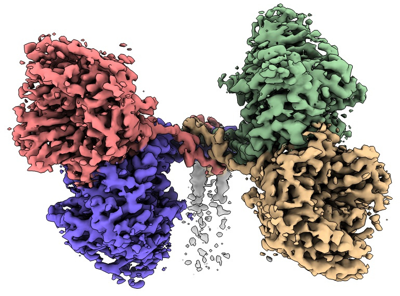
| Title | Cryo-EM structure of the canine distemper virus tetrameric attachment glycoprotein | |||||||||
 Map data Map data | final sharpened map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | canine distemper virus / morbillivirus cell entry / receptor-binding protein H / soluble H ectodomain / VIRAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationhost cell membrane / host cell surface receptor binding / viral envelope / symbiont entry into host cell / virion attachment to host cell / virion membrane Similarity search - Function | |||||||||
| Biological species |  Canine morbillivirus Canine morbillivirus | |||||||||
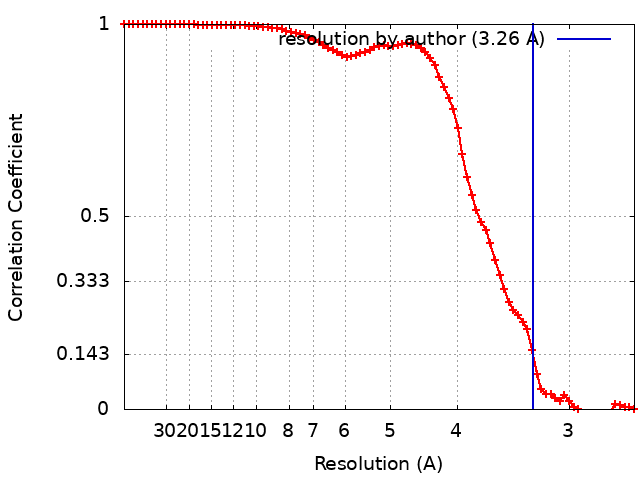
| Method | single particle reconstruction / cryo EM / Resolution: 3.26 Å | |||||||||
 Authors Authors | Kalbermatter D / Jeckelmann J-M / Wyss M / Plattet P / Fotiadis D | |||||||||
| Funding support |  Switzerland, 1 items Switzerland, 1 items
| |||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2023 Title: Structure and supramolecular organization of the canine distemper virus attachment glycoprotein. Authors: David Kalbermatter / Jean-Marc Jeckelmann / Marianne Wyss / Neeta Shrestha / Dimanthi Pliatsika / Rainer Riedl / Thomas Lemmin / Philippe Plattet / Dimitrios Fotiadis / Abstract: Canine distemper virus (CDV) is an enveloped RNA morbillivirus that triggers respiratory, enteric, and high incidence of severe neurological disorders. CDV induces devastating outbreaks in wild and ...Canine distemper virus (CDV) is an enveloped RNA morbillivirus that triggers respiratory, enteric, and high incidence of severe neurological disorders. CDV induces devastating outbreaks in wild and endangered animals as well as in domestic dogs in countries associated with suboptimal vaccination programs. The receptor-binding tetrameric attachment (H)-protein is part of the morbilliviral cell entry machinery. Here, we present the cryo-electron microscopy (cryo-EM) structure and supramolecular organization of the tetrameric CDV H-protein ectodomain. The structure reveals that the morbilliviral H-protein is composed of three main domains: stalk, neck, and heads. The most unexpected feature was the inherent asymmetric architecture of the CDV H-tetramer being shaped by the neck, which folds into an almost 90° bent conformation with respect to the stalk. Consequently, two non-contacting receptor-binding H-head dimers, which are also tilted toward each other, are located on one side of an intertwined four helical bundle stalk domain. Positioning of the four protomer polypeptide chains within the neck domain is guided by a glycine residue (G158), which forms a hinge point exclusively in two protomer polypeptide chains. Molecular dynamics simulations validated the stability of the asymmetric structure under near physiological conditions and molecular docking showed that two receptor-binding sites are fully accessible. Thus, this spatial organization of the CDV H-tetramer would allow for concomitant protein interactions with the stalk and head domains without steric clashes. In summary, the structure of the CDV H-protein ectodomain provides new insights into the morbilliviral cell entry system and offers a blueprint for next-generation structure-based antiviral drug discovery. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_14842.map.gz | 40.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-14842-v30.xmlemd-14842.xml | 20.7 KB 20.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_14842_fsc.xml | 8.5 KB | Display | FSC data file |
| Images |  emd_14842.png emd_14842.png | 185.6 KB | ||
| Masks | emd_14842_msk_1.map | 42.9 MB | Mask map | |
| Filedesc metadata | emd-14842.cif.gz | 6.3 KB | ||
| Others | emd_14842_additional_1.map.gzemd_14842_half_map_1.map.gzemd_14842_half_map_2.map.gz | 21.5 MB 39.8 MB 39.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-14842ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14842 http://ftp.pdbj.org/pub/emdb/structures/EMD-14842ftp://ftp.pdbj.org/pub/emdb/structures/EMD-14842 | HTTPS FTP |
-Related structure data
| Related structure data |  7znyMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |
-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_14842.map.gz / Format: CCP4 / Size: 42.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | final sharpened map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.29171 Å | ||||||||||||||||||||||||||||||||||||
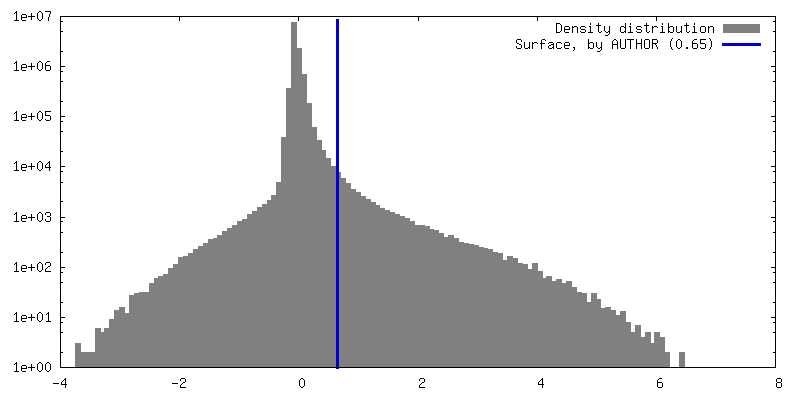
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















-Supplemental data
-Mask #1
| File | emd_14842_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
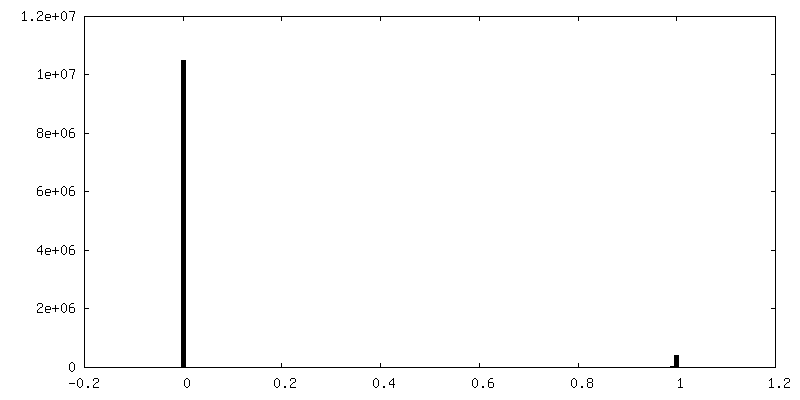
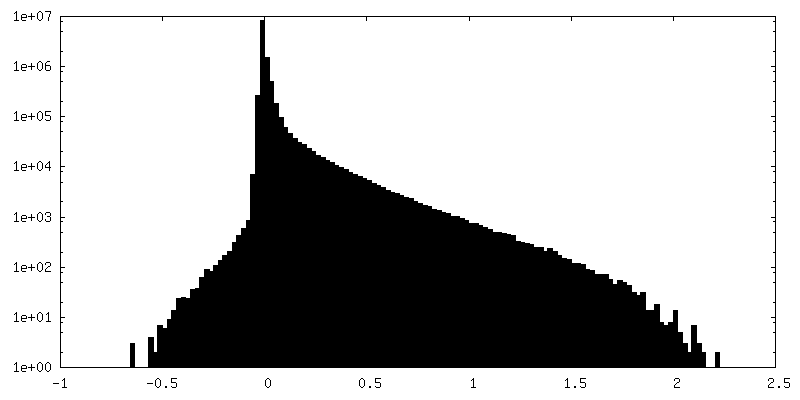
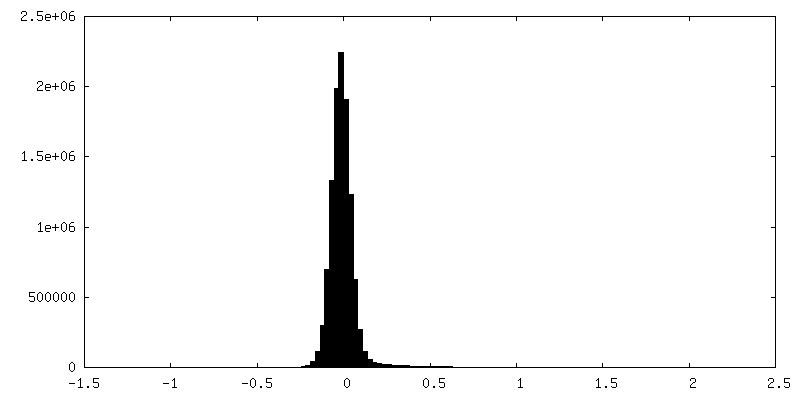
| Density Histograms |






-Additional map: full map
| File | emd_14842_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | full map | ||||||||||||
| Projections & Slices |
| ||||||||||||
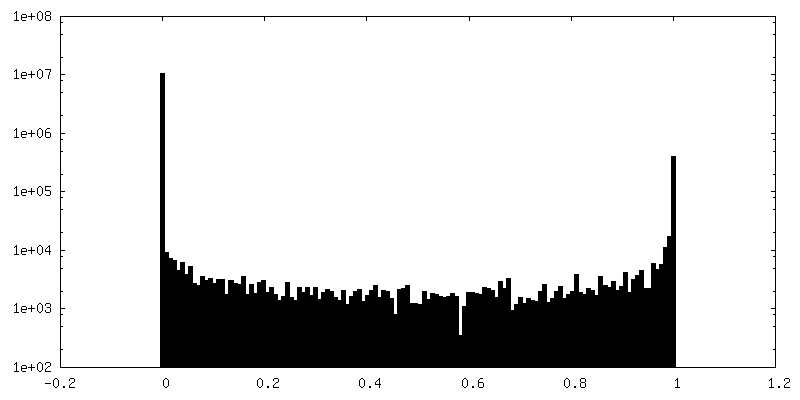
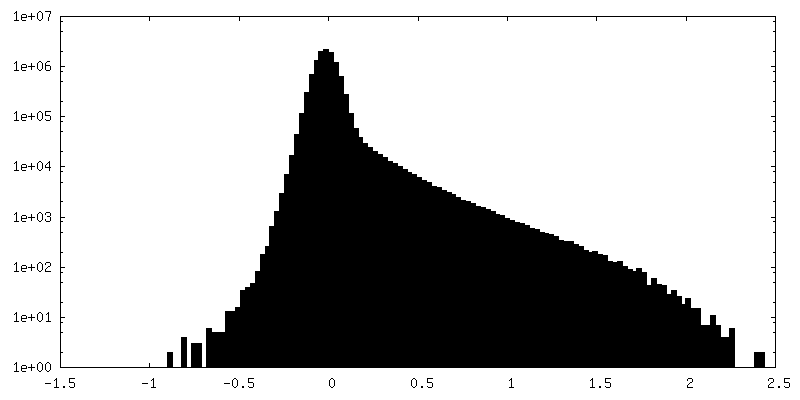
| Density Histograms |






-Half map: half map1
| File | emd_14842_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
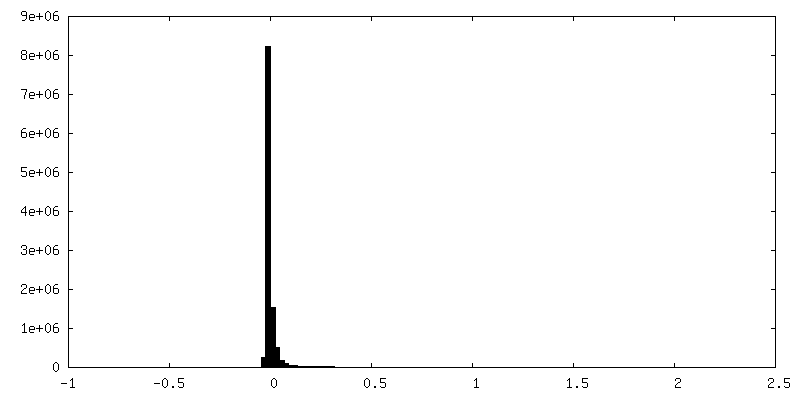
| Density Histograms |



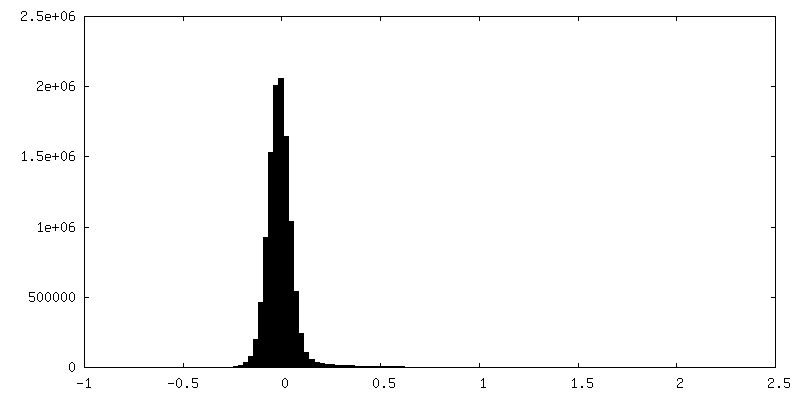
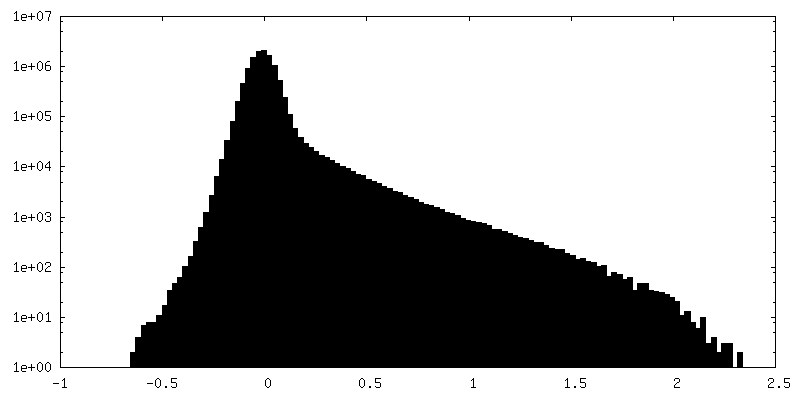
-Half map: half map2
| File | emd_14842_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



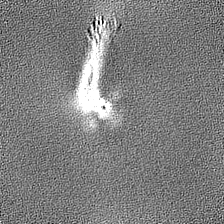

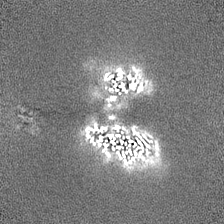
- Sample components
Sample components
-Entire : Canine morbillivirus
| Entire | Name: Canine morbillivirus |
|---|---|
| Components |
|
-Supramolecule #1: Canine morbillivirus
| Supramolecule | Name: Canine morbillivirus / type: virus / ID: 1 / Parent: 0 / Macromolecule list: all / NCBI-ID: 11232 / Sci species name: Canine morbillivirus / Virus type: VIRUS-LIKE PARTICLE / Virus isolate: OTHER / Virus enveloped: Yes / Virus empty: Yes |
|---|---|
| Molecular weight | Theoretical: 272 KDa |
-Macromolecule #1: Hemagglutinin glycoprotein
| Macromolecule | Name: Hemagglutinin glycoprotein / type: protein_or_peptide / ID: 1 / Number of copies: 4 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Canine morbillivirus |
| Molecular weight | Theoretical: 68.311031 KDa |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Sequence | String: MLSYQDKVSA FYKDNARANS SKLSLVTEEQ GGRRPPYLLF VLLILLVGIM ALLAITGVRF HQVSTSNMEF SRLLKEDMEK SEAVHHQVI DVLTPLFKII GDEIGLRLPQ KLNEIKQFIL QKTNFFNPNR EFDFRDLHWC INPPSKIKVN FTNYCDTIGI R KSIASAAN ...String: MLSYQDKVSA FYKDNARANS SKLSLVTEEQ GGRRPPYLLF VLLILLVGIM ALLAITGVRF HQVSTSNMEF SRLLKEDMEK SEAVHHQVI DVLTPLFKII GDEIGLRLPQ KLNEIKQFIL QKTNFFNPNR EFDFRDLHWC INPPSKIKVN FTNYCDTIGI R KSIASAAN PILLSALSGG RGDIFPPYRC SGATTSVGRV FPLSVSLSMS LISRTSEIIN MLTAISDGVY GKTYLLVPDY IE GGFDTQK IRVFEIGFIK RWLNDMPLLQ TTNYMVLPEN SKAKVCTIAV GELTLASLCV DESTVLLYHD SDGSQDGILV VTL GIFGAT PMDQVEEVIP VAHPSVEKIH ITNHRGFIKD SIATWMVPAL VSEKQEEQKN CLESACQRKS YPMCNQTSWE PFGG GQLPS YGRLTLPLDP SIDLQLNISF TYGPVILNGD GMDYYESPLL DSGWLTIPPK NGTVLGLINK ASRGDQFTVI PHVLT FAPR ESSGNCYLPI QTSQIMDKDV LTESNLVVLP TQNFRYVIAT YDISRGDHAI VYYVYDPIRA ISYTYPFRLT TKGRPD FLR IECFVWDDDL WCHQFYRFEA DSTNSTTSVE NLVRIRFSCN RSKP UniProtKB: Hemagglutinin glycoprotein |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.1 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 200 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 120 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.025 kPa | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV Details: Blot Time: 3.5 sec Blot Force: -6 Drain Time: 0 Wait Time: 0. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 2 / Number real images: 26835 / Average exposure time: 1.5 sec. / Average electron dose: 50.0 e/Å2 Details: Two Datasets from two different grids were recorded: 13,760 and 13,075 movies |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 20.0 µm / Nominal defocus min: 8.0 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: RIGID BODY FIT |
|---|---|
| Output model | PDB-7zny: |
